July 12, 2024
IDSA is committed to providing up-to-date guidance on the treatment of antimicrobial-resistant (AMR) infections. This fourth updated guidance document focuses on infections caused by extended-spectrum β-lactamase-producing Enterobacterales (ESBL-E), AmpC β- lactamase-producing Enterobacterales (AmpC-E), carbapenem-resistant Enterobacterales (CRE), Pseudomonas aeruginosa with difficult-to-treat resistance (DTR P. aeruginosa), carbapenem-resistant Acinetobacter baumannii (CRAB), and Stenotrophomonas maltophilia. This updated document replaces previous versions of the guidance document.
Please submit your feedback and comments on the AMR Guidance by emailing us at PracticeGuidelines@idsociety.org.
Published by CID, August 7, 2024 | Clinical Infectious Diseases, ciae403, https://doi.org/10.1093/cid/ciae403
Pranita D. Tamma*, Emily L. Heil, Julie Ann Justo, Amy J. Mathers, Michael J. Satlin, & Robert A. Bonomo,
*Corresponding Author: Pranita D. Tamma, MD, MHS, Johns Hopkins University School of Medicine, Department of Pediatrics, Baltimore, Maryland, USA; ptamma1@jhmi.edu
Keywords: ESBL; AmpC; carbapenem-resistant Enterobacterales; Pseudomonas aeruginosa; CRAB; Stenotrophomonas maltophilia
Update History
July 18, 2023
Version 3.0 of the guidance has been released.
Notable Updates from the 2023 IDSA AMR Guidance Document
The reader is encouraged to review the entire AMR Guidance document as edits and updated pre-clinical and clinical data have been made to virtually all questions. The following represent some key changes to the 2023 IDSA AMR Guidance Document.
ESBL-E
- Fosfomycin continues to not be suggested for pyelonephritis and complicated urinary tract infections (cUTI); however, the uncertainty of the additive benefit of additional doses of oral fosfomycin for these indications was highlighted in light of recent clinical data.
- Amoxicillin-clavulanic acid continues to not be a preferred agent for uncomplicated ESBL-producing cystitis; however, it was acknowledged that there may be occasions where it is prescribed if resistance or toxicities preclude the use of alternative oral antibiotics and there is a preference to avoid IV antibiotics. It is advised that caution be given to patients about the potential increased risk of recurrent infection if amoxicillin-clavulanic acid is administered for this indication.
- Additional details on the mechanistic reasons why piperacillin-tazobactam is not anticipated to be effective for ESBL-E infections are provided.
- Piperacillin-tazobactam continues to not be preferred for the treatment of pyelonephritis and cUTI; however, it was acknowledged that if piperacillin-tazobactam was initiated for pyelonephritis or cUTI caused and clinical improvement occurs, the decision to continue piperacillin-tazobactam should be made with the understanding that theoretically there may be an increased risk for microbiological failure with this approach.
- A re-review of available data and newer data indicate that ceftolozane-tazobactam is likely to be effective against ESBL-E; however, it suggested that this agent be preserved for the treatment of DTR aeruginosa or polymicrobial infections (e.g., both DTR P. aeruginosa and ESBL-E).
AmpC-E
- The term “moderate to high risk” clinically significant AmpC production was replaced with “moderate risk” throughout.
- It was clarified that even without upregulation of AmpC production, basal production of AmpC β-lactamases by organisms with inducible ampC expression leads to intrinsic resistance to ampicillin, amoxicillin-clavulanate, ampicillin-sulbactam, and first- and second-generation cephalosporins.
- The suggestion that cefepime is not advised for Enterobacter cloacae, Citrobacter freundii, and Klebsiella aerogenes with cefepime MICs of 4-8 µg/mL because of concerns for an increased risk of ESBL production in this cefepime MIC range was removed in light of newer data and a rereview of existing data.
CRE
- An increase in the prevalence of CRE isolates producing metallo-beta-lactamases (MBL) in the United States (e.g., NDM, VIM, IMP) is acknowledged.
- A description of a Clinical and Laboratory Standards Institute (CLSI) endorsed method (i.e., broth disk elution method) to test for activity of the combination of ceftazidime-avibactam and aztreonam for MBL-producing Enterobacterales is discussed.
- Dosing suggestions for ceftazidime-avibactam in combination with aztreonam are updated in Table 1 and Supplemental Material. Both agents are suggested to be administered every 8 hours to facilitate simultaneous administration in clinical practice.
DTR P. aeruginosa
- For infections caused by P. aeruginosa isolates not susceptible to any carbapenem agent but susceptible to traditional β-lactams (e.g., cefepime), administration of a traditional agent as high-dose extended-infusion therapy continues to be suggested, although the panel no longer emphasizes the importance of repeating AST on the initial isolate before administration of the traditional agent given the frequency with which this susceptibility profile occurs.
- A new question (i.e., Question 4.2) has been added “Are there differences in percent activity against DTR aeruginosa across available β-lactam agents?” Differences in DTR P. aeruginosa susceptibility percentages to the newer β-lactams are described along with regional differences in enzymatic mechanisms of resistance that contribute to some of these differences.
- Once-daily tobramycin or amikacin were added as alternative treatment options for pyelonephritis or cUTI caused by DTR aeruginosa given the prolonged duration of activity of these agents in the renal cortex and the convenience of once daily dosing.
CRAB
- Sulbactam-durlobactam, in combination with meropenem or imipenem-cilastatin, was added as the preferred agent for the treatment of CRAB infections.
- High-dose ampicillin-sulbactam in combination with at least one other agent has been changed from a preferred to an alternative regimen if sulbactam-durlobactam is not available.
- The suggested dosing of high-dose ampicillin-sulbactam has been adjusted to be 27 grams of ampicillin-sulbactam (18 grams ampicillin, 9 grams sulbactam) daily.
S. maltophilia
- Questions have been adjusted to list agents in order of preference (i.e., cefiderocol [with a second agent at least initially], ceftazidime-avibactam and aztreonam, minocycline [with a second agent], TMP-SMX [with a second agent], or levofloxacin [with a second agent].
- A description of a CLSI endorsed method (i.e., broth disk elution method) to test for activity of the combination of ceftazidime-avibactam and aztreonam for maltophilia activity is discussed.
- Tigecycline has been removed as a component of combination therapy.
- Updated guidance from the CLSI advising against the testing of ceftazidime for maltophilia infections has been added.
Abstract
Background: The Infectious Diseases Society of America (IDSA) is committed to providing up-to-date guidance on the treatment of antimicrobial-resistant (AMR) infections. This guidance document focuses on infections caused by extended-spectrum β-lactamase-producing Enterobacterales (ESBL-E), AmpC β- lactamase-producing Enterobacterales (AmpC-E), carbapenem-resistant Enterobacterales (CRE), Pseudomonas aeruginosa with difficult-to-treat resistance (DTR P. aeruginosa), carbapenem-resistant Acinetobacter baumannii (CRAB), and Stenotrophomonas maltophilia. This updated document replaces previous versions of the guidance document.
Methods: A panel of six infectious diseases specialists with expertise in managing antimicrobial- resistant infections formulated questions about the treatment of infections caused by ESBL-E, AmpC-E, CRE, DTR P. aeruginosa, CRAB, and S. maltophilia. Because of differences in the epidemiology of AMR and availability of specific anti-infectives internationally, this document focuses on the treatment of AMR infections in the United States.
Results: Preferred and alternative suggested treatment approaches are provided with accompanying rationales, assuming the causative organism has been identified and antibiotic susceptibility results are known. Approaches to empiric treatment, transitioning to oral therapy, duration of therapy, and other management considerations are discussed briefly. Suggested approaches apply for both adult and pediatric populations, although suggested antibiotic dosages are provided only for adults.
Conclusions: The field of AMR is highly dynamic. Consultation with an infectious diseases specialist is recommended for the treatment of AMR infections. This document is current as of December 31, 2023 and will be updated periodically. The most current version of this document, including date of publication, is available at www.idsociety.org/practice-guideline/amr-guidance/.
Introduction
Antimicrobial-resistant (AMR) infections are a global crisis. Internationally, approximately 1.3 million deaths were estimated to be directly attributable to AMR pathogens in 20191. In the United States, AMR pathogens caused more than 2.8 million infections and over 35,000 deaths annually from 2012 through 2017, according to the Centers for Disease Control and Prevention (CDC) Antibiotic Resistance Threats in the United States Report2.
As an alternative to practice guidelines, the Infectious Diseases Society of America (IDSA) has endorsed developing more narrowly focused guidance documents for the treatment of infections where data may not be very robust and continue to rapidly evolve – such as with AMR. Guidance documents are prepared by a small team of experts, who answer questions about treatment based on a comprehensive (but not necessarily systematic) review of the literature, clinical experience, and expert opinion. Documents are made available online and updated annually.
In the present document, guidance is provided on the treatment of infections caused by extended-spectrum β-lactamase-producing Enterobacterales (ESBL-E), AmpC β-lactamase-producing Enterobacterales (AmpC-E), carbapenem-resistant Enterobacterales (CRE), Pseudomonas aeruginosa with difficult-to-treat resistance (DTR P. aeruginosa), carbapenem-resistant Acinetobacter baumannii (CRAB), and Stenotrophomonas maltophilia. Many of these pathogens have been designated urgent or serious threats by the CDC2. Each pathogen causes a wide range of infections that are encountered in United States hospitals of all sizes, and that carry with them significant morbidity and mortality.
Guidance is presented in the form of answers to a series of clinical questions for each pathogen.
Although brief descriptions of notable clinical trials, resistance mechanisms, and antimicrobial susceptibility testing (AST) methods are included, the document does not provide a comprehensive review of these topics. GRADE methodology (i.e., Grading of Recommendations, Assessment, Development, and Evaluations) are not employed. Due to differences in the molecular epidemiology of resistance and availability of specific antibiotics internationally, treatment suggestions are geared toward AMR infections in the United States. This guidance document applies to both adult and pediatric populations. Suggested antibiotic dosing for adults with AMR infections, assuming normal renal and hepatic function, are provided in Table 1. Pediatric dosing is not provided. The content of this document is current as of December 31, 2023. The most current version of this IDSA guidance document and corresponding date of publication is available at: www.idsociety.org/practice-guideline/amr-guidance.
General Management Recommendations
Suggested treatment approaches in this guidance document assume that the causative organism has been identified and that in vitro activity of antibiotics is demonstrated. If two antibiotics are equally effective, important considerations in selecting a specific agent include safety, cost, convenience, and local formulary availability.
Complicated Urinary Tract Infection Definition
In this document, the term cUTI refers to UTIs occurring in association with a structural or functional abnormality of the genitourinary tract, or any UTI in an adolescent or adult male. In general, the panel suggests cUTI be treated with similar agents and for similar treatment durations as pyelonephritis. For cUTI where the source has been controlled (e.g., removal of a Foley catheter) and ongoing concerns for urinary stasis or indwelling urinary hardware are no longer present, it is reasonable to select antibiotic agents and treatment durations similar to those that would be selected for uncomplicated cystitis, with day 1 of therapy being the day source control occurred.
Empiric Therapy
Empiric treatment decisions are outside the scope of this guidance document. However, in general, empiric therapy should be informed by the most likely pathogens, severity of illness of the patient, the likely source of the infection, and any additional patient-specific factors (e.g., severe penicillin allergy, severe immune compromise, chronic kidney disease). When determining empiric treatment for a given patient, clinicians should also consider: (1) previous organisms identified from the patient and associated antimicrobial susceptibility testing (AST) data in the last 12 months3, (2) antibiotic exposure within the past 3 months3, and (3) local AST patterns for the most likely pathogens. Treatment decisions should be refined based on the identity and the AST profile of the pathogen, as well as on the identification of any prominent β-lactamase genes that have been identified.
For all organisms, but for DTR P. aeruginosa, CRAB, and S. maltophilia in particular, a distinction between bacterial colonization and infection is important because unnecessary antibiotic therapy will only further the development of resistance and may cause unnecessary antibiotic related harm to patients. Commonly selected empiric antibiotic regimens are generally not active against CRAB and S. maltophilia infections. The decision to target treatment for CRAB and/or S. maltophilia in empiric antibiotic regimens should involve a careful risk-benefit analysis after reviewing previous culture results, clinical presentation, individual host risk factors, and antibiotic-specific adverse event profiles.
Duration of Therapy and Transitioning to Oral Therapy
Recommendations on durations of therapy are not provided, but clinicians are advised that the duration of therapy should not differ for infections caused by organisms with resistant phenotypes compared to infections caused by more susceptible phenotypes4. After AST results are available, it may become apparent that inactive antibiotic therapy was initiated empirically. This may impact the duration of therapy. For example, uncomplicated cystitis is typically a mild infection 5. If an antibiotic not active against the causative organism was administered empirically for uncomplicated cystitis, but clinical improvement nonetheless occurred, it is generally not necessary to repeat a urine culture, change the antibiotic regimen, or extend the planned treatment course. However, for all other infections, if AST results indicate a potentially inactive agent was initiated empirically, a change to an active regimen for a full treatment course (dated from the start of active therapy) is suggested. Additionally, important host factors related to immune status, ability to attain source control, and general response to therapy should be considered when determining treatment durations for AMR infections, as with the treatment of any bacterial infection. Finally, whenever possible, transitioning to oral therapy should be considered (assuming IV therapy was initially prescribed), particularly if the following criteria are met: (1) susceptibility to an appropriate oral agent is demonstrated, (2) the patient is hemodynamically stable (3) reasonable source control measures have occurred, and (4) concerns about insufficient intestinal absorption are not present 6.
Table 1 and Table 2
Table 1. Suggested dosing of antibiotics for the treatment of antimicrobial-resistant infections in adults, assuming
normal renal and hepatic function1,2
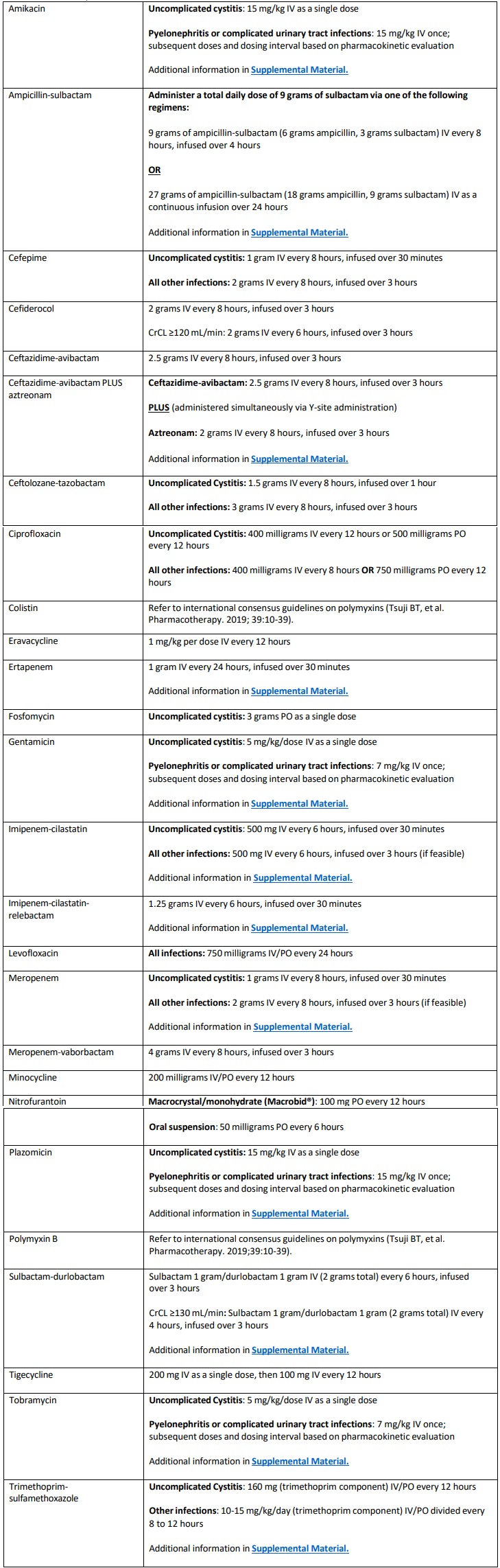
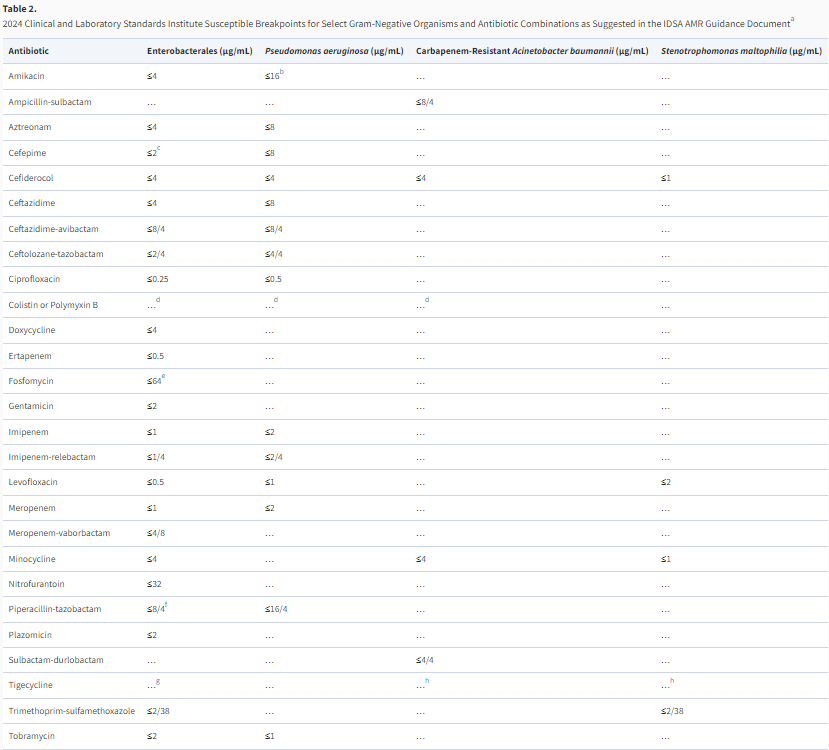
Table 2. 2024 Clinical and Laboratory Standards Institute Breakpoints for Select Gram-Negative Organisms and
Antibiotic Combinations as Suggested in the IDSA AMR Guidance Document1
Section 1: Extended-spectrum β-lactamase-Producing Enterobacterales
ESBLs are enzymes that inactivate most penicillins, cephalosporins, and aztreonam. EBSL-E generally remain susceptible to carbapenems. ESBLs do not inactivate non-β-lactam agents (e.g., ciprofloxacin, trimethoprim-sulfamethoxazole [TMP-SMX], gentamicin). However, organisms carrying ESBL genes often harbor additional genes or mutations in genes expanding their resistance to a broad range of antibiotics.
Any gram-negative organism has the potential to harbor ESBL genes; however, they are most prevalent in Escherichia coli, Klebsiella pneumoniae, Klebsiella oxytoca, and Proteus mirabilis7-9. CTX-M enzymes, particularly CTX-M-15, are the most common ESBLs in the United States9. ESBLs other than CTX-M with unique hydrolyzing abilities are also present, including variants of TEM and SHV β- lactamases with amino acid substitutions, but they have undergone less rigorous clinical investigation than CTX-M enzymes10-14. Routine EBSL testing is not performed by most clinical microbiology laboratories15,16. Rather, non-susceptibility to ceftriaxone (i.e., ceftriaxone minimum inhibitory concentrations [MICs] ≥2 µg/mL), is often used as a proxy for ESBL production, although this threshold has limitations with specificity as organisms not susceptible to ceftriaxone for reasons other than ESBL production may be falsely presumed to be ESBL-producers17,18. For this guidance document, ESBL-E refers to presumed or confirmed ESBL-producing E. coli, K. pneumoniae, K. oxytoca, or P. mirabilis. Treatment suggestions for ESBL-E infections assume that in vitro activity of preferred and alternative antibiotics has been demonstrated.
Question 1.1: What are preferred antibiotics for the treatment of uncomplicated cystitis caused by ESBL-E?
Suggested approach: Nitrofurantoin and TMP-SMX are preferred treatment options for uncomplicated cystitis caused by ESBL-E. Ciprofloxacin, levofloxacin, and carbapenems are alternative agents for uncomplicated cystitis caused by ESBL-E. Although effective, their use is discouraged when nitrofurantoin or TMP-SMX are active. An aminoglycoside (as a single dose) and oral fosfomycin (for E. coli only) are also alternative treatments for uncomplicated cystitis caused by ESBL-E.
Rationale
Nitrofurantoin and TMP-SMX have been shown to be effective options for uncomplicated cystitis, including uncomplicated ESBL-E cystitis5,19,20 21. Although carbapenems and the fluoroquinolones ciprofloxacin or levofloxacin are effective agents against ESBL-E cystitis22,23, their use for uncomplicated cystitis is discouraged when other effective options are available. Limiting use of these agents preserves their activity for future infections when treatment options may be more restricted. Moreover, limiting their use reduces the risk of associated toxicities, particularly with the fluoroquinolones, which have been associated with an increased risk for prolonged QTc intervals, tendinitis and tendon rupture, aortic dissections, seizures, peripheral neuropathy, and Clostridioides difficile infections24-27.
Treatment with a single intravenous (IV) dose of an aminoglycoside is an alternative treatment option for uncomplicated ESBL-E cystitis. Aminoglycosides are nearly exclusively eliminated by the renal route. A single IV dose is generally effective for uncomplicated cystitis, with minimal toxicity, but robust clinical trial data are lacking28. Oral fosfomycin is an alternative treatment option exclusively for uncomplicated ESBL-E cystitis caused by E. coli. Susceptibility of E. coli to fosfomycin is not routinely tested by most clinical microbiology laboratories but E. coli resistance to fosfomycin remains rare in the United States29,30. CLSI breakpoints are only available for E. coli for fosfomycin. Fosfomycin is not suggested for the treatment of infections caused by K. pneumoniae and several other gram-negative organisms which frequently carry fosA hydrolase genes that may lead to clinical failure31,32. A randomized open-label trial indicated that a single dose of oral fosfomycin is associated with higher clinical failure than a five-day course of nitrofurantoin for uncomplicated cystitis 19. Although this trial was not limited to E. coli cystitis, in a subgroup analysis exclusively of E. coli infections, outcomes remained poor in the fosfomycin group with day 14 clinical failure of 50% in the fosfomycin group versus 22% in the nitrofurantoin group19. The additive benefit of additional doses of oral fosfomycin for uncomplicated cystitis is not known but may be a reasonable option as has been suggested for cUTI33 (Question 1.2).
Amoxicillin-clavulanic is not suggested for the treatment of ESBL-E cystitis. A randomized clinical trial compared a three-day regimen of amoxicillin-clavulanic acid (500 mg/125 mg twice daily) to a three-day course of ciprofloxacin (250 mg twice daily) for 370 women with uncomplicated E. coli cystitis22. Clinical cure was observed in 58% and 77% of the women randomized to the amoxicillin- clavulanic and ciprofloxacin arms, respectively. The higher failure rates with amoxicillin-clavulanic acid appear to be associated with persistent vaginal bacterial colonization, which occurred in 45% and 10% of patients in the amoxicillin-clavulanic acid and ciprofloxacin arms, respectively22. The proportion of women in the trial infected with ESBL-E strains is not available. Of note, both agents were administered at dosages lower than generally suggested (Table 1). Even though data indicate that clavulanic acid is effective against ESBLs in vitro34,35, this may not translate to clinical efficacy36. Robust data indicating that oral amoxicillin-clavulanic acid is effective for ESBL-E uncomplicated cystitis are lacking. While amoxicillin-clavulanic acid is not a preferred agent for uncomplicated ESBL-producing cystitis, if it is prescribed because resistance or toxicities preclude use of alternative oral antibiotics and there is a preference to avoid IV antibiotics, caution should be given to patients about the potential increased risk of recurrent infection if amoxicillin-clavulanic acid is administered.
The panel suggests avoiding doxycycline for the treatment of ESBL-E uncomplicated cystitis. Two clinical outcomes studies, published nearly 50 years ago, demonstrated that oral tetracyclines may be effective for the treatment of UTIs37,38. Both of these studies, however, primarily focused on P. aeruginosa, an organism not susceptible to oral tetracyclines, questioning the impact that antibiotic therapy had on clinical cure. Doxycycline is primarily eliminated through the intestinal tract39 with limited urinary excretion (35-60%)39. Until more convincing data demonstrating the clinical effectiveness of oral doxycycline for the treatment of ESBL-E cystitis are available, the panel suggests against the use of doxycycline for this indication. The roles of piperacillin-tazobactam, cefepime, and the cephamycins for the treatment of uncomplicated cystitis are discussed in Question 1.4, Question 1.5, and Question1.6, respectively.
Question 1.2: What are preferred antibiotics for the treatment of pyelonephritis or cUTI caused by ESBL-E?
Suggested approach: TMP-SMX, ciprofloxacin, or levofloxacin are preferred treatment options for pyelonephritis or cUTIs caused by ESBL-E. Ertapenem, meropenem, and imipenem-cilastatin are preferred agents when resistance or toxicities preclude the use of TMP-SMX or fluoroquinolones. Aminoglycosides are alternative options for the treatment of ESBL-E pyelonephritis or cUTI.
Rationale
TMP-SMX, ciprofloxacin, and levofloxacin are preferred treatment options for patients with ESBL-E pyelonephritis or cUTIs, assuming in vitro susceptibility has been demonstrated, based on the ability of these agents to achieve adequate and sustained concentrations in the urine, clinical trial results, and clinical experience40-42. Carbapenems are also preferred agents, when resistance or toxicities prevent the use of TMP-SMX or fluoroquinolones, or early in the treatment course if a patient is critically ill (Question 1.3). If a carbapenem is initiated and susceptibility to TMP-SMX, ciprofloxacin, or levofloxacin is demonstrated, transitioning to oral formulations of these agents is preferred over completing a treatment course with a carbapenem. Limiting use of carbapenem exposure will preserve their activity for future AMR infections, which frequently often arise in patients with cUTIs43.
Aminoglycosides are alternative options for pyelonephritis and cUTI. Although expected to be effective, they are considered alternative agents because of their associated nephrotoxicity risk. Animal models suggest aminoglycosides concentrate in the renal parenchyma44. In a clinical trial of 609 adults receiving plazomicin for cUTI infections, clinical relapse occurred in 2% versus 7% and increases in serum creatinine levels of ≥0.5 mg above baseline occurred in 7% versus 4% of patients in the plazomicin and meropenem groups, respectively45. In general, higher percentages of Enterobacterales clinical isolates are susceptible to plazomicin compared to other aminoglycosides46. Other aminoglycosides are likely equally effective for the treatment of ESBL-E pyelonephritis or cUTI if susceptibility is demonstrated45,47,48. Of note, in 2023 the CLSI revised gentamicin, tobramycin, and amikacin breakpoints for the Enterobacterales16 (Table 2). Aminoglycosides may be reasonable to consider for completing treatment courses (e.g., transitioning from another agent for terminal doses) given their prolonged duration of activity in the renal cortex and the convenience of once daily dosing47,48 (Table 1, Supplemental Material). Duration-dependent risks of nephrotoxicity should be considered with all aminoglycosides49,50.
Fosfomycin is not suggested for the treatment of pyelonephritis or cUTI given its limited renal parenchymal concentrations. More data are needed to evaluate the role of oral fosfomycin for patients with pyelonephritis or cUTI, particularly when administered as a multidose regimen and after several days of preferred therapy. In a clinical trial of 97 women with E. coli pyelonephritis (approximately half of patients had associated bacteremia) who received up to 5 days of IV therapy, participants were subsequently transitioned to either once-daily 3 g doses of oral fosfomycin or twice daily 500 mg doses of oral ciprofloxacin for 10 days of total antibiotic therapy51. Similar clinical cure percentages were identified in both groups (75% versus 65%, respectively). However, only approximately 6% of isolates were ESBL-producing, limiting generalizability to pyelonephritis caused by drug-resistant phenotypes51. Moreover, as 7 days is generally sufficient for the treatment of pyelonephritis, the attributable benefit of the additional days of oral fosfomycin or ciprofloxacin is unclear. Another clinical trial randomized 51 patients with cUTI to 3 g of fosfomycin daily or 750 mg of levofloxacin daily for 5-7 days, after up to two days of IV therapy33. Clinical cure at the end of therapy was similar in both treatment groups (69% versus 68%). In this study, 63% of infections were caused by E. coli but only one isolate in each arm was caused by an ESBL-producing isolate.
IV fosfomycin is not clinically available in the United States. Although some data suggest IV fosfomycin may have activity against organisms beyond E. coli, it is difficult to translate data from IV fosfomycin to oral fosfomycin given the limited oral bioavailability and lower daily dosages with oral fosfomycin52. Transitioning to daily oral fosfomycin needs further investigation before suggesting for or against this practice for the treatment of ESBL-E pyelonephritis or cUTI; however, it may be a reasonable option when other preferred or alternative oral options are not available.
Fosfomycin is an alternative option for the treatment of prostatitis caused by ESBL-producing E. coli when preferred options (i.e., carbapenems, TMP-SMX, or fluoroquinolones) cannot be tolerated or do not test susceptible53-59. In an observational study, fosfomycin, dosed at 3 g orally daily for one week, followed by 3 g orally every 48 hours for 6 to 12 weeks, was associated with clinical cure in 36 (82%) of 44 males with chronic bacterial prostatitis53. Fosfomycin is not suggested for prostatitis caused by gram- negative organisms other than E. coli due to the likely presence of the fosA gene and its ability to inactive this agent (Question 1.1).
Nitrofurantoin does not achieve adequate concentrations in the renal parenchyma and is not advised for the treatment of pyelonephritis or cUTI. Doxycycline is also not advised for the treatment of ESBL-E pyelonephritis or cUTIs due to its limited urinary excretion (Question 1.1)39. The roles of piperacillin-tazobactam, cefepime, and the cephamycins for the treatment of pyelonephritis or cUTIs are discussed in Question 1.4, Question 1.5, and Question 1.6, respectively.
Question 1.3: What are preferred antibiotics for the treatment of infections outside of the urinary tract caused by ESBL-E?
Suggested approach: Meropenem, imipenem-cilastatin, or ertapenem are preferred for the treatment of infections outside of the urinary tract caused by ESBL-E. For patients who are critically ill and/or experiencing hypoalbuminemia, meropenem or imipenem-cilastatin are the preferred carbapenems. After appropriate clinical response is achieved, transitioning to oral TMP-SMX, ciprofloxacin, or levofloxacin should be considered, if susceptibility is demonstrated.
Rationale
A carbapenem is recommended as first-line treatment of ESBL-E infections outside of the urinary tract, based primarily on data from a large clinical trial, as described below60. Meropenem, imipenem-cilastatin, or ertapenem are preferred agents; ertapenem offers a more convenient option for patients needing to continue carbapenem therapy in the outpatient setting when oral treatment options are not available.
For patients who are critically ill and/or experiencing hypoalbuminemia, meropenem or imipenem-cilastatin are the preferred carbapenems. Ertapenem, in contrast to meropenem and imipenem, is highly protein bound leading to a relatively prolonged serum half-life61. In patients with hypoalbuminemia, the free fraction of ertapenem increases, leading to increased ertapenem clearance and a significant decrease in the serum half-life of this agent, which may not be optimal with daily dosing of this agent62-64. An observational study of 279 patients with Enterobacterales infections found that hypoalbuminemia (defined as serum albumin <2.5 g/dL) was associated with an approximately five- times higher odds of 30-day mortality for patients receiving ertapenem compared to those receiving meropenem or imipenem-cilastatin65. Clinical literature regarding the use of ertapenem, relative to other carbapenems, in critically ill patients is limited and conflicting64,66. However, given known pharmacokinetic (PK) alterations in patients with critical illness and limitations in the pharmacokinetic and pharmacodynamic (PK/PD) profile of ertapenem67,68, the panel suggests the use of meropenem or imipenem-cilastatin, rather than ertapenem, as initial therapy in critically ill patients with ESBL-E infections. Higher doses of ertapenem (e.g., 1.5 grams) or more frequent dosing (e.g., every 12 hours) may circumvent some of the probability of target attainment issues with ertapenem in obese and critically ill patients with hypoalbuminemia, respectively, but data for these alternative dosing strategies are limited67,69-71.
The clinical trial which established carbapenem therapy as the treatment of choice for ESBL-E bloodstream infections randomized 391 patients with ceftriaxone non-susceptible E. coli or K. pneumoniae (87% later confirmed to have ESBL genes) to piperacillin-tazobactam 4.5 g IV every six hours or meropenem 1 g IV every eight hours, both as standard infusions (i.e., over 30 minutes). The primary outcome of 30-day mortality occurred in 12% and 4% of patients receiving piperacillin- tazobactam and meropenem, respectively60. Trial data were subsequently reanalyzed only including patients with clinical isolates against which piperacillin-tazobactam MICs were ≤16 µg/mL by broth microdilution, the reference standard for AST72. Reanalyzing the data from 320 (82%) patients with clinical isolates available for retesting, 30-day mortality occurred in 9% versus 4% of those in the piperacillin-tazobactam and meropenem arms, respectively. Although the absolute risk difference was attenuated and no longer significant in the reanalysis (i.e., the 95% confidence interval ranged from −1% to 11%)72, the panel still suggests carbapenem therapy as the preferred treatment of ESBL-producing bloodstream infections due to the notable direction of the risk difference. Limitations of piperacillin-tazobactam are further described in Question 1.4. Comparable clinical trial data are not available for
ESBL-E infections from other body sites. Nevertheless, the panel suggests extrapolating evidence for ESBL-E bloodstream infections to other common sites of infection, such as intra-abdominal infections, skin and soft tissue infections, and pneumonia. Similarly, although the trial evaluated meropenem, the panel suggests extending the findings to imipenem-cilastatin and ertapenem, with the latter limited to patients with normal serum albumin and patients who are not critically ill.
Data from observational studies support the use of oral step-down therapy for Enterobacterales bloodstream infections, including those caused by AMR isolates, after appropriate clinical milestones are achieved73,74. Based on the high bioavailability and sustained serum concentrations of oral TMP-SMX and fluoroquinolones, these agents should be treatment considerations for patients with ESBL-E infections if (1) susceptibility to one of these agents is demonstrated, (2) the patient is hemodynamically stable, (3) reasonable source control has occurred, and (4) concerns about insufficient intestinal absorption are not present6.
Clinicians should avoid oral step-down to nitrofurantoin, fosfomycin, amoxicillin-clavulanic acid, omadacycline, or doxycycline for ESBL-E bloodstream infections. Nitrofurantoin and fosfomycin achieve poor serum concentrations. Amoxicillin-clavulanic acid, omadacycline, and doxycycline have limited data to support their use for ESBL-E bloodstream infections.
Question 1.4: Is there a role for piperacillin-tazobactam in the treatment of infections caused by ESBL- E?
Suggested approach: If piperacillin-tazobactam was initiated as empiric therapy for uncomplicated cystitis caused by an organism later identified as an ESBL-E and clinical improvement occurs, no change or extension of antibiotic therapy is necessary. The panel suggests TMP-SMX, ciprofloxacin, levofloxacin, or carbapenems rather than piperacillin-tazobactam for the treatment of ESBL-E pyelonephritis or cUTI, with the understanding that the risk of clinical failure with piperacillin-tazobactam may be low. Piperacillin-tazobactam is not suggested for the treatment of infections outside of the urinary tract caused by ESBL-E, even if susceptibility to piperacillin-tazobactam is demonstrated.
Rationale
Piperacillin-tazobactam often demonstrates in vitro activity against ESBL-E75. However, there are several concerns regarding tazobactam’s ability to function as an effective β-lactamase inhibitor. First, piperacillin-tazobactam MIC testing may be inaccurate and/or poorly reproducible when ESBL enzymes are present, or in the presence of other β-lactamase enzymes such as OXA-1, making it unclear if an isolate that tests susceptible to this agent is reliably susceptible 72,76-79. Second, in vitro data indicate that with increased bacterial inoculum which may be present in certain clinical infections (e.g., abscesses), regrowth of ESBL-E isolates appears significantly more likely in the setting of piperacillin-tazobactam compared with meropenem; the clinical implications of these findings are unclear80-82. Third, the effectiveness of tazobactam may be diminished for organisms with increased expression of ESBL enzymes or by the presence of multiple ESBL or other β-lactamases (e.g., AmpC enzymes)83. This may in part be due to the low concentration of tazobactam relative to the amount of piperacillin. As an example, in a 4.5 g dose of piperacillin-tazobactam there is an 8:1 ratio of piperacillin to tazobactam (i.e., 4 grams of piperacillin and 0.5 grams of tazobactam). In contrast, in a 3 g dose of ceftolozane there is a 2:1 ratio of ceftolozane to tazobactam. It is plausible that the lower dose of tazobactam in piperacillin-tazobactam may limit its abilities as an inhibitor84. Finally, the piperacillin-tazobactam breakpoint for Enterobacterales is primarily based on PK/PD considerations of piperacillin dosing strategies and not on whether a fixed concentration of 4 µg/ml of tazobactam in testing wells is reflective of the restorative ability of common tazobactam dosages to reestablish the activity of piperacillin in the setting of ESBL enzymes84,85.
If piperacillin-tazobactam was initiated as empiric therapy for uncomplicated cystitis caused by an organism later identified as an ESBL-E and clinical improvement occurs, no change or extension of antibiotic therapy is necessary, as uncomplicated cystitis often resolves on its own. Determining the role of piperacillin-tazobactam for the treatment of ESBL-E pyelonephritis or cUTI is a more challenging. Several observational studies have found similar clinical outcomes when comparing the efficacy of piperacillin-tazobactam and carbapenems for the treatment of ESBL-E pyelonephritis or cUTI 86-88 89,90. A randomized, open-label clinical trial investigating this question was also conducted 91. The trial included 66 patients with ESBL-producing E. coli pyelonephritis or cUTI (with confirmation of the presence of ESBL genes) randomized to either piperacillin-tazobactam 4.5 g IV every six hours or ertapenem 1 g IV every 24 hours. Clinical success was similar between the groups at 94% for piperacillin-tazobactam and 97% for ertapenem. These studies suggest non-inferiority between piperacillin-tazobactam and carbapenems for pyelonephritis or cUTIs. In the subgroup of 231 patients with ESBL-E bloodstream infections from a urinary source in the aforementioned clinical trial (Question 1.3), higher mortality was identified in the piperacillin-tazobactam group (7% vs 3%) 60, although not achieving statistical significance. Evaluating the totality of the data, the panel prefers carbapenem therapy (or oral trimethoprim-sulfamethoxazole, ciprofloxacin, or levofloxacin, if susceptible) for the treatment of ESBL-E pyelonephritis or cUTIs, but acknowledges it is may be reasonable to prescribe piperacillin-tazobactam for these infections based on the results of available comparative effectiveness studies (Question 1.2). If piperacillin-tazobactam was initiated as empiric therapy for pyelonephritis or cUTI caused by an organism later identified as an ESBL- E and clinical improvement occurs, the decision to continue piperacillin-tazobactam should be made with the understanding that theoretically there may be an increased risk for microbiological failure with this approach.
Observational studies have had conflicting results regarding the effectiveness of piperacillin- tazobactam for the treatment of ESBL-E bloodstream infections86-88,91-103. A clinical trial of ESBL-E bloodstream infections indicated inferior results with piperacillin-tazobactam compared to carbapenem therapy (Question 1.3) 60. A second trial investigating the role of piperacillin-tazobactam for the treatment of ESBL-E bloodstream infections is ongoing104.
In 2022, the CLSI lowered the piperacillin-tazobactam breakpoints for the Enterobacterales. MICs of ≤8/4 µg/mL are considered susceptible and an MIC of 16 µg/mL is considered susceptible, dose- dependent (Table 2)105. In the clinical trial mentioned in Question 1.3, 94% of isolates would have been considered susceptible or susceptible dose-dependent to piperacillin-tazobactam if applying the revised piperacillin-tazobactam breakpoints, indicating that in the presence of ESBL production, susceptibility to piperacillin-tazobactam may not correlate with clinical success60,72.
Question 1.5: Is there a role for cefepime in the treatment of infections caused by ESBL-E?
Suggested approach: If cefepime was initiated as empiric therapy for uncomplicated cystitis caused by an organism later identified as an ESBL-E and clinical improvement occurs, no change or extension of antibiotic therapy is necessary. The panel suggests avoiding cefepime for the treatment of pyelonephritis or cUTI. Cefepime is also not suggested for the treatment of infections outside of the urinary tract caused by ESBL-E, even if susceptibility to cefepime is demonstrated.
Rationale
ESBLs commonly hydrolyze cefepime83,106. Furthermore, even if ESBL-producing isolates test susceptible to cefepime, cefepime MIC testing may be inaccurate and/or poorly reproducible with commercial AST methods107. Clinical trials designed to compare the outcomes of patients with ESBL-E bloodstream infections treated with cefepime or carbapenem have not been conducted.
If cefepime was initiated as empiric therapy for uncomplicated cystitis caused by an organism later identified as an ESBL-E and clinical improvement occurs, no change or extension of antibiotic therapy is necessary, as uncomplicated cystitis often resolves on its own. Limited data are available evaluating the role of cefepime versus carbapenems for ESBL-E pyelonephritis and cUTI91,108 89. A clinical trial evaluating the treatment of molecularly confirmed ESBL-E pyelonephritis and cUTI terminated the cefepime arm early because of a high clinical failure signal with cefepime (2 g IV every 12 hours), despite all isolates having cefepime MICs of 1-2 µg/mL91. Until more robust comparative effectiveness studies are available to inform the role of cefepime, the panel suggests avoiding cefepime for the treatment of ESBL-E pyelonephritis or cUTI.
Clinical trials comparing cefepime to carbapenems for ESBL-E bloodstream infections have not been conducted. However, a randomized trial comparing cefepime (2 g IV every 8 hours) to imipenem- cilastatin (500 mg IV every 6 hours) for nosocomial pneumonia identified clinical failure in 4 of 13 patients (31%) with pneumonia due to ESBL-E in the cefepime arm, compared to none of 10 patients (0%) in the imipenem-cilastatin arm109. Observational studies that compare cefepime and carbapenems for the treatment of invasive ESBL-E infections demonstrated either no difference in outcomes or poorer outcomes with cefepime 110-113. For these reasons, the panel suggests avoiding cefepime for the treatment of invasive ESBL-E infections.
Question 1.6: Is there a role for the cephamycins in the treatment of infections caused by ESBL-E?
Suggested approach: Cephamycins are not suggested for the treatment of ESBL-E infections until more clinical outcomes data using cefoxitin or cefotetan are available and optimal dosing has been defined.
Rationale
The cephamycins are cephalosporins that are generally able to withstand hydrolysis from ESBL enzymes114,115. The cephamycins available in the United States are cefoxitin and cefotetan which are both IV agents. At least ten observational studies have compared the clinical outcomes of patients with ESBL-E infections—generally UTIs or bloodstream infections from urinary sources—treated with cephamycins versus carbapenems 116-125. Eight of the ten investigations found no difference in clinical outcomes116,118-120,122,123; two studies demonstrated poorer outcomes with cephamycins117,121. One of the two studies included 57 patients with K. pneumoniae bloodstream infections; 14-day mortality was 55% and 39% in the cephamycin and carbapenem arms, respectively117. The second study was the largest published to date, including 380 patients with E. coli and K. pneumoniae bloodstream infections; 30-day mortality was 29% versus 13% in the cephamycin (i.e., floxomef) and carbapenem arms, respectively 121. Importantly, all eight studies were observational, included diverse sources of infection, had notable selection bias, and used a variety of cephamycins with differences in dosing, duration, and frequency of administration.
The panel does not suggest cephamycins for the treatment of ESBL-E infections, including ESBL-E uncomplicated cystitis. Many of the cephamycins investigated in observational studies are not available in the United States. Limited numbers of patients received cefoxitin or cefotetan in published studies 119,123,126. The panel believes more clinical data associated with these agents for the treatment of ESBL-E infections are necessary before advocating for their use—including optimal dosing and frequency of administration. Data suggest more favorable outcomes with high-dose, continuous infusion cefoxitin (i.e., 6 g per day infused continuously) 123,126, but this is challenging to administer. As both cefotetan and cefoxitin are only available IV and have relatively short half-lives, there does not appear to be a feasibility advantage with use of these agents over preferred agents for the treatment of ESBL-E infections.
Question 1.7: What is the role of newer β-lactam-β-lactamase inhibitor combinations and cefiderocol for the treatment of infections caused by ESBL-E?
Suggested approach: The panel suggests that ceftazidime-avibactam, meropenem-vaborbactam, imipenem-cilastatin-relebactam, ceftolozane-tazobactam, and cefiderocol be preferentially reserved for treating infections caused by organisms exhibiting carbapenem resistance.
Rationale
Ceftazidime-avibactam, meropenem-vaborbactam, imipenem-cilastatin-relebactam, ceftolozane-tazobactam, and cefiderocol exhibit activity against ESBL-E127-129. Avibactam is able to successfully protect ceftazidime against hydrolysis by binding to and inhibiting the function of ESBL enzymes9,130. Subgroup analysis of clinical trial data support ceftazidime-avibactam effectiveness against ESBL-E infections131-135.
The carbapenem component of meropenem-vaborbactam and imipenem-cilastatin-relebactam provide sufficient activity against ESBL-E, even without the addition of a β-lactamase inhibitor.
Ceftolozane-tazobactam appears more potent against ESBL-E than piperacillin-tazobactam with ceftolozane MICs reducing several dilutions lower than piperacillin MICs, with the addition of tazobactam136-141. Moreover, ceftolozane appears to have greater stability to hydrolysis by common ESBL enzymes (e.g., CTX-M-15) compared to piperacillin, making ceftolozane less reliant than piperacillin on tazobactam's inhibitory properties142,143. Additionally, the ratio of β-lactam to tazobactam present in ceftolozane-tazobactam (2:1) results in greater concentration of tazobactam compared to piperacillin- tazobactam (8:1).
In a subgroup analysis of 72 patients with ESBL-E intra-abdominal infections in a randomized clinical trial, ceftolozane-tazobactam was associated with similar clinical cure as meropenem144. In a randomized clinical trial comparing ceftolozane-tazobactam versus meropenem for pneumonia, 28-day mortality in the subgroup of patients with ESBL-E pneumonia was similar between the 84 patients receiving ceftolozane-tazobactam (21%) and the 73 patients receiving meropenem (29%)145,146. Clinical cure and microbiologic eradication rates were also similar between the ceftolozane-tazobactam and meropenem arms.
Although ceftazidime-avibactam, meropenem-vaborbactam, imipenem-cilastatin-relebactam, ceftolozane-tazobactam, and cefiderocol are expected to be effective against ESBL-E infections, the panel suggests that these agents be preferentially reserved for treating carbapenem-resistant organisms or polymicrobial infections including organisms exhibiting carbapenem resistance (e.g., ceftolozane- tazobactam for coinfection with DTR P. aeruginosa and ESBL-E).
Section 2: AmpC β-Lactamase-Producing Enterobacterales
AmpC β-lactamases are enzymes that are produced at basal levels by a number of Enterobacterales and glucose non-fermenting gram-negative organisms. Their primary function is to assist with cell wall recycling147. AmpC β-lactamases are capable of hydrolyzing a number of β-lactam agents (to a level that makes the agents ineffective), some in settings of basal AmpC production (e.g., cefazolin) and others in settings of increased AmpC production (e.g., ceftriaxone). Increased AmpC production by Enterobacterales generally occurs by one of three mechanisms: (1) inducible chromosomal gene expression, (2) stable chromosomal gene de-repression, or (3) constitutively expressed ampC genes (frequently carried on plasmids, but sometimes integrated into the bacterial chromosome)147-149.
Increased AmpC enzyme production resulting from inducible ampC expression can occur in the presence of specific antibiotics and results in sufficient AmpC enzyme in the periplasmic space to increase MICs to certain antibiotics (i.e., ceftriaxone, cefotaxime, ceftazidime, aztreonam, and piperacillin-tazobactam). In this scenario, an Enterobacterales isolate that initially tests susceptible to ceftriaxone may exhibit non-susceptibility to this agent after treatment with ceftriaxone is initiated. In this guidance document, such organisms are described as having a moderate risk for clinically significant AmpC production. Resistance due to ampC induction can be observed after even a few doses of ceftriaxone, cefotaxime, or ceftazidime 150.
For the other two mechanisms (i.e., stable chromosomal de-repression or constitutively overexpressed ampC genes), AmpC production is always increased. Isolates with either of these two mechanisms are expected to test non-susceptible to ceftriaxone, cefotaxime, and/or ceftazidime. As such, infections by organisms with these resistance mechanisms generally pose less of a treatment dilemma than infections caused by isolates with inducible ampC expression. Regarding the first of these two mechanisms, some Enterobacterales isolates (e.g., certain Escherichia coli and Shigella spp.) contain mutations in promoters or attenuators of ampC or other related genes (e.g., ampD, ampR, ampG), stably de-repressing gene expression 151. For the second mechanism, constitutive expression of ampC genes (e.g., blaCMY, blaFOX, blaDHA, blaACT, blaMIR) occurs 152. These ampC genes can be found either on plasmids (e.g., blaCMY in E. coli) or be integrated into the bacterial chromosome (e.g., blaCMY in Citrobacter freundii). In this document, we will focus on the treatment of infections by Enterobacterales species with a moderate likelihood of inducible ampC gene expression (i.e., the first of the three mechanisms)153,154.
Question 2.1: Which commonly identified Enterobacterales species should be considered at moderate risk for clinically significant inducible ampC production?”
Suggested approach: Enterobacter cloacae complex, Klebsiella aerogenes, and Citrobacter freundii are the most common Enterobacterales at moderate risk for clinically significant inducible AmpC production.
Rationale
Quantifying the likelihood of ampC induction across bacterial species would be best defined by systematically identifying organisms initially susceptible to certain β-lactam agents (e.g., ceftriaxone) that, on subsequent isolation (and after β-lactam exposure), become resistant, with genotyping and expression studies to confirm that the same organism was recovered and that AmpC production significantly increased. Unfortunately, such studies are not available.
Commonly used acronyms to denote organisms at risk for AmpC production (e.g., SPACE, SPICE, ESCPM) obscure the wide range of ampC induction potential among gram-negative organisms and ignore variance within bacterial genera 147,148. For example, C. freundii harbors a chromosomal ampC whereas Citrobacter koseri does not 155-157. Thus, current acronyms may be overly simplistic and associated with both an “undercalling” and “overcalling” of the likelihood of clinically significant AmpC production among individual bacterial species. As another example, “indole positive Proteus species” are often included in existing acronyms. Indole-positive Proteus spp. currently refers to organisms such as P. vulgaris, which generally does not contain a chromosomal ampC gene. The terminology “indole positive Proteus species” previously included Proteus rettgeri and Proteus morganii (since renamed Providencia rettgeri and Morganella morganii, respectively) 158, making the inclusion of “indole-positive Proteus spp.” in mnemonics for organisms at moderate risk of AmpC production no longer accurate.
The emergence of clinically relevant ampC expression during antibiotic treatment has been most frequently described for E. cloacae complex (herein, referred to as E. cloacae for simplicity), K. aerogenes (formerly Enterobacter aerogenes), and C. freundii. Clinical reports suggest that the emergence of resistance after exposure to an agent like ceftriaxone may occur in approximately 20% of infections caused by these organisms150,159-163. These clinical observations mirror in vitro mutation rate analyses, which also suggest that these organisms are likely to overexpress ampC 164. Therefore, when E. cloacae, K. aerogenes, or C. freundii are recovered in clinical cultures (other than urine cultures in uncomplicated cystitis), the panel suggests generally avoiding treatment with ceftriaxone, cefotaxime, or ceftazidime, even if an isolate initially tests susceptible to these agents (Question 2.2). Even without upregulation of AmpC production, basal production of AmpC β-lactamases in these organisms leads to intrinsic resistance to ampicillin, amoxicillin-clavulanate, ampicillin-sulbactam, and first- and second- generation cephalosporins16.
In contrast, other organisms historically presumed to be at moderate risk for the development of clinically significant ampC expression, such as Serratia marcescens, Morganella morganii, and Providencia spp., are significantly less likely to overexpress ampC based on both in vitro analysis 164,165 and clinical reports 150,159,166. Available data suggest that clinically significant AmpC production occurs in less than 5% of these organisms. When S. marcescens, M. morgannii, or Providencia spp. are recovered from clinical cultures, the panel suggests selecting antibiotic treatment according to AST results. Basal production of AmpC β-lactamase renders these organisms intrinsically resistant to ampicillin, amoxicillin-clavulanate, and first- and second-generation cephalosporins16.
A number of less common clinical pathogens (e.g., Hafnia alvei, Citrobacter youngae, Yersinia enterocolitica) that carry inducible chromosomal ampC genes have not undergone significant investigation 164,167-169. As such, descriptions of their potential for clinically significant AmpC production are very limited. It is reasonable to use AST results to guide treatment decisions if these organisms are recovered in clinical cultures (e.g., administer ceftriaxone if susceptible to ceftriaxone). When treating infections caused by these less commonly recovered organisms (or caused by S. marcescens, M. morgannii, or Providencia spp.) with a high bacterial burden and limited source control (e.g., endocarditis, central nervous system infections), it is alternatively reasonable to consider treatment with cefepime instead of ceftriaxone, even if the organism tests susceptible to ceftriaxone. As with all infections, if an adequate clinical response is not observed after appropriately dosed antibiotic therapy is initiated and necessary source control measures are taken, clinicians should consider the possibility of the emergence of resistance to the initially prescribed agent.
Question 2.2: What features should be considered in selecting antibiotics for infections caused by organisms at moderate risk of clinically significant AmpC production due to an inducible ampC gene?
Suggested approach: Several β-lactam antibiotics are at moderate risk of inducing ampC genes. Both the ability to induce ampC genes and the relative stability of the agent against hydrolysis by AmpC should inform antibiotic decision-making.
Rationale
β-lactam antibiotics fall within a spectrum of potential for inducing ampC genes. Aminopenicillins (i.e., amoxicillin, ampicillin), narrow-spectrum (i.e., first generation) cephalosporins, and cephamycins are potent ampC inducers 170,171. However, both organisms at low risk (e.g., S. marcescens) and at moderate risk (e.g., E. cloacae) for clinically significant ampC induction hydrolyze these antibiotics even at basal ampC expression levels. Therefore, such AmpC-E isolates will generally test as resistant to these drugs or are not recommended to be tested due to intrinsic resistance, averting treatment dilemmas. Imipenem is also a potent ampC inducer but it generally remains stable to AmpC-E hydrolysis because of the formation of stable acyl enzyme complexes 170. The induction potential of ertapenem and meropenem has not been formally investigated but, similar to imipenem, they are generally stable to AmpC hydrolysis 172,173. Ceftriaxone, cefotaxime, ceftazidime, piperacillin-tazobactam, and aztreonam are relatively weak ampC inducers 171,174. Available evidence indicates that despite their limited ability to induce ampC, the susceptibility of these agents to hydrolysis makes them less likely to be effective for the treatment of infections by organisms at moderate risk for clinically significant AmpC production173,175-177. They remain, however, reasonable treatment agents for Enterobacterales at lower risk for clinically significant AmpC production (e.g., S. marcescens).
Cefepime has the advantage of both being a weak inducer of ampC and of withstanding hydrolysis by AmpC β-lactamases because of the formation of stable acyl enzyme complexes 178,179. Therefore, cefepime is generally an effective agent for the treatment of AmpC-E infections 180. TMP- SMX, fluoroquinolones, aminoglycosides, tetracyclines, and other non-β-lactam antibiotics do not induce ampC and are also not substrates for AmpC hydrolysis.
Question 2.3: What is the role of cefepime for the treatment of infections caused by Enterobacterales at moderate risk of clinically significant AmpC production due to an inducible ampC gene?
Suggested approach: Cefepime is suggested for the treatment of infections caused by organisms at moderate risk of significant AmpC production (i.e., E. cloacae complex, K. aerogenes, and C. freundii).
Rationale
Cefepime is an oxyimino-cephalosporin that is relatively stable against AmpC enzymes and that also has low ampC induction potential 178,179,181,182. Clinical trials comparing clinical outcomes of patients with AmpC-E infections treated with cefepime versus carbapenem therapy are not available. However, several observational studies suggest cefepime is associated with similar clinical outcomes as carbapenem therapy 163,183,184 185,186. Furthermore, a meta-analysis including seven studies comparing clinical outcomes of patients receiving cefepime versus carbapenems for Enterobacter spp., Citrobacter spp., and Serratia spp. bloodstream infections did not find differences in clinical outcomes between these treatment regimens 180. However, considerable heterogeneity between studies existed, ill- appearing patients were more likely to receive carbapenem therapy, and risk of clinically significant AmpC production varied by the included species. In light of both the advantages of cefepime as a compound and no clear clinical failure signals in the literature when administered for the treatment of AmpC-E infections, the panel suggests cefepime as a preferred treatment option for E. cloacae, K. aerogenes, and C. freundii infections (Table 1).
Although cefepime may be effective for the treatment of AmpC-E infections, it remains suboptimal against infections caused by ESBL-E, which is a consideration if both enzymes may be produced by an Enterobacterales (Question 1.5). In a study from Taiwan, 89% of E. cloacae isolates with cefepime MICs of 4-8 µg/mL (i.e., susceptible dose-dependent) were ESBL-producing 111. The same study evaluated 217 patients with E. cloacae bloodstream infections and found that all 10 patients with infections caused by ESBL-producing isolates with cefepime MICs of 4-8 µg/mL who received cefepime died within 30 days. In contrast, none of the six patients who received cefepime for infections caused by non-ESBL-producing cefepime isolates with MICs of 4-8 µg/mL died within 30 days 111.
Data are incomplete on the frequency of ESBL production by Enterobacterales at moderate risk of clinically significant AmpC production in the United States. An evaluation of 211 consecutive E. cloacae isolates from 66 United States hospitals from 2019-2020 indicated that 3% contained a blaCTX-M gene8. A study from Pittsburgh found that 15 of 45 (33%) E. cloacae bloodstream isolates collected between 2003-2005 produced SHV-type ESBLs187. There was no association between ESBL production and the cefepime MIC. A study from Baltimore found that ESBL genes were identified in 22% of K. aerogenes (4/18), 14% of E. cloacae (7/51), and in no C. freundii (0/8 [0%]) bloodstream isolates collected between 2018-2021188. There was no correlation between the presence of an ESBL gene and the cefepime MIC; none of the ESBL-producing isolates had cefepime MICs of 4-8 µg/mL188.
Contemporary data specific to the United States are needed to better understand how frequently ESBLs are produced by Enterobacterales at moderate risk of clinically significant AmpC production. Available data do not suggest there is a clear association between cefepime susceptible dose-dependent MICs (i.e., MICs 4-8 µg/mL) and ESBL production16. Cefepime susceptible dose-dependent MICs are based on cefepime dosages of 2 grams every 8 hours, infused over 3 hours and this dosing strategy is suggested to treat Enterobacterales infections with cefepime MICs in this range186 (Table 2).
Question 2.4: What is the role of ceftriaxone for the treatment of infections caused by Enterobacterales at moderate risk of clinically significant AmpC production due to an inducible ampC gene?
Suggested approach: Ceftriaxone (or cefotaxime or ceftazidime) is not suggested for the treatment of invasive infections caused by organisms at moderate risk of clinically significant AmpC production (e.g., E. cloacae complex, K. aerogenes, and C. freundii). Ceftriaxone is reasonable for uncomplicated cystitis caused by these organisms when susceptibility is demonstrated.
Rationale
Clinical reports differ on how frequently resistance to ceftriaxone emerges during the treatment of infections by Enterobacterales at moderate risk for clinically significant ampC induction. Several challenges exist when interpreting studies that have attempted to address this question. First, there are no CLSI-endorsed approaches for AmpC detection in clinical isolates, making quantifying their production difficult. Second, these organisms may display ceftriaxone resistance for other reasons (e.g., ESBL production); however, such mechanisms are rarely investigated in clinical studies for organisms other than E. coli, K. pneumoniae, K. oxytoca, and P. mirabilis. Third, studies often combine estimates for organisms at low risk for significant AmpC production (e.g., S. marcescens, M. morgannii) with those posing a higher risk (e.g., E. cloacae, C. freundii), obscuring an understanding of how frequently resistance to ceftriaxone emerges for organisms at moderate risk for clinically significant AmpC production 189. Fourth, studies that evaluate the proportion of isolates exhibiting ceftriaxone non-susceptibility after ceftriaxone exposure do not include confirmation of genetic relatedness of index and subsequent isolates. Additionally, many AmpC clinical studies used pre-2010 CLSI ceftriaxone breakpoints (i.e., ceftriaxone MICs ≤8 µg/mL), making translation of prevalence estimates to current CLSI ceftriaxone susceptibility breakpoints of ≤1 µg/mL challenging 16,189. Finally, in addition to selection bias, there is significant heterogeneity in sources of infections, severity of illness, pre-existing medical conditions, co-administration of additional antibiotics, and ceftriaxone dosing and duration across studies, complicating the interpretation of clinical data.
These limitations notwithstanding, available data suggest that the emergence of resistance after ceftriaxone exposure occurs in approximately 20% of infections caused by E. cloacae, K. aerogenes, or C. freundii 150,159-163,190-192. Comparative effectiveness studies addressing the management of presumed AmpC-producing infections have mostly focused on the emergence of ceftriaxone resistance, rather than on clinical outcomes. No clinical trials have compared the outcomes of patients with presumed AmpC-E infections treated with ceftriaxone compared to alternate agents (e.g., cefepime). A number of observational studies compared the clinical outcomes of patients with infections caused by E. cloacae, K. aerogenes, and C. freundii treated with ceftriaxone compared with other β-lactams160,190,191,193-197. Most of these studies did not identify differences in clinical outcomes when comparing patients treated with ceftriaxone versus carbapenems, with the limitations outlined above.
Nonetheless, since available data indicate a reasonable risk for the emergence of resistance when ceftriaxone (or other third-generation cephalosporins) is prescribed for infections caused by organisms at moderate risk of AmpC production (i.e., infections caused by E. cloacae, K. aerogenes, C. freundii), the panel suggests generally avoiding third-generation cephalosporins when treating infections caused by these organisms. Based on the mild nature of uncomplicated cystitis and the sufficient urinary excretion of ceftriaxone, ceftriaxone may be adequate therapy for the management of AmpC-E uncomplicated cystitis. For other relatively uncomplicated infections it may be reasonable to transition to ceftriaxone after clear clinical improvement has been achieved and if there are no concerns for ongoing sources of infection (e.g., abscesses, indwelling catheters), weighing the convenience of once- daily ceftriaxone dosing with the potentially increased risk of emergence of resistance.
Question 2.5: What is the role of piperacillin-tazobactam for the treatment of infections caused by Enterobacterales at moderate risk of clinically significant AmpC production due to an inducible ampC gene?
Suggested approach: Piperacillin-tazobactam is not suggested for the treatment of invasive infections caused by Enterobacterales at moderate risk of clinically significant inducible AmpC production.
Rationale
Tazobactam is less effective at protecting β-lactams from AmpC hydrolysis than newer β- lactamase inhibitors, such as avibactam, relebactam, and vaborbactam149,173,174,198. The role of piperacillin-tazobactam in treating Enterobacterales at moderate risk for clinically significant AmpC production remains uncertain. A 2019 meta-analysis summarized the findings of eight observational studies and did not identify a difference in mortality between patients treated with piperacillin- tazobactam and carbapenems for bacteremia caused by Enterobacter spp., Citrobacter spp., or Serratia spp. 189 However, significant heterogeneity across studies and confounding by indication likely existed (i.e., ill appearing patients were more likely to be prescribed carbapenems). In two observational studies included in this meta-analysis, 30-day mortality among patients treated with piperacillin-tazobactam was numerically higher than for patients treated with carbapenems (15% [6/41 patients] versus 7% [3/41 patients] 199 and 45% [10/22 patients] versus 11% [5/45 patients], respectively) 194. At least two other observational studies including 103 and 81 patients, respectively, with bloodstream infections caused by Enterobacterales known to harbor chromosomal ampC genes indicated significantly poorer clinical outcomes for patients treated with piperacillin-tazobactam compared with cefepime or carbapenem therapy192,200.
A pilot unblinded clinical trial compared the outcomes of 72 patients with bloodstream infections caused by Enterobacter spp., K. aerogenes, C. freundii, M. morganii, Providencia spp., or S. marcescens randomized to piperacillin-tazobactam (4.5 grams IV every 6 hours as a standard infusion) or meropenem (1 gram IV every 8 hours as a standard infusion) 201. There were no significant differences in the primary outcome (a composite outcome including 30-day mortality, clinical failure, microbiological failure, or microbiological relapse) between the study arms. However, some notable and seemingly conflicting findings were observed for individual components of this composite outcome: mortality (0% versus 6%, p=0.13); clinical failure (21% versus 12%, p=0.29); microbiological failure (13% versus 0%), p=0.03), and microbiological relapse (0% versus 9%, p=0.06), for the piperacillin-tazobactam and meropenem arms, respectively. The findings of this trial are challenging to interpret and a larger trial is needed to more definitively determine the role of piperacillin-tazobactam for the treatment of organisms at moderate risk for clinically significant ampC induction.
In light of the limited ability of tazobactam to protect piperacillin from AmpC hydrolysis in vitro and at least four observational studies identifying poorer clinical outcomes in patients prescribed piperacillin-tazobactam 191,194,199,200, the panel suggests against prescribing piperacillin-tazobactam for serious infections caused by AmpC-E.
Piperacillin-tazobactam may be a reasonable treatment option for mild infections such as uncomplicated cystitis – although narrower-spectrum agents are generally preferred. For other relatively uncomplicated infections it may be reasonable to transition to piperacillin-tazobactam in settings of adverse events to preferred agents (e.g., neurotoxicity associated with cefepime) or other patient-specific factors (e.g., polymicrobial infections), after considering the potentially increased risk of treatment failure with piperacillin-tazobactam therapy. This practice is only advised after clinical improvement has been achieved and if there are no concerns for ongoing sources of infection (e.g., abscesses, indwelling catheters).
Question 2.6: What is the role of newer β-lactam-β-lactamase inhibitor combinations and cefiderocol for the treatment of infections caused by Enterobacterales at moderate risk of clinically significant AmpC production due to an inducible ampC gene?
Suggested approach: The panel suggests that ceftazidime-avibactam, meropenem-vaborbactam, imipenem-cilastatin-relebactam, and cefiderocol be preferentially reserved for treating infections caused by organisms exhibiting carbapenem resistance. The panel does not suggest the use of ceftolozane-tazobactam as a treatment option for AmpC-E infections.
Rationale
Ceftazidime-avibactam, meropenem-vaborbactam, and imipenem-cilastatin-relebactam generally exhibit in vitro activity against AmpC-E 130,202-204. Ceftazidime-avibactam is likely to be effective as a treatment for infections caused by AmpC-E205. Although the frequency is unknown, emergence of resistance of AmpC-E to ceftazidime-avibactam has been described, generally due to amino acid changes in the omega loop region of the AmpC enzyme206-208. Carbapenems are generally stable to hydrolysis by AmpC-E; by extension meropenem-vaborbactam and imipenem-cilastatin-relebactam are expected to be effective treatment options for AmpC-E.
Cefiderocol demonstrates in vitro activity against AmpC-E 129,209 and it is likely to be effective in clinical practice, although some case reports indicate the potential for AmpC-E to develop resistance to this agent 206,207. Although ceftazidime-avibactam, meropenem-vaborbactam, imipenem-cilastatin- relebactam, and cefiderocol are likely to be effective against AmpC-E infections, the panel suggests that these agents be preferentially reserved for treating infections caused by organisms exhibiting carbapenem resistance, where a greater need for them exists.
Ceftolozane was developed to be more resistant to hydrolysis than earlier cephalosporins against Pseudomonas-derived AmpC cephalosporinases; however, less is known about ceftolozane- tazobactam’s activity against AmpC-E. Tazobactam is less effective at protecting β-lactams from AmpC hydrolysis compared with newer β-lactamase inhibitors, such as avibactam, relebactam, and vaborbactam149,173,174,198. While some in vitro data suggest ceftolozane-tazobactam has activity against AmpC-E 210, in at least one investigation the agent was active against only 19% of E. cloacae isolates producing moderate levels of AmpC enzymes211. Clinical outcomes data for ceftolozane-tazobactam for the treatment of AmpC-E infections are limited; a clinical trial evaluating this question is underway 212. Based on the limited available data, the panel suggests against the use of ceftolozane-tazobactam as a treatment option for AmpC-E infections.
In polymicrobial infections in which DTR P. aeruginosa and AmpC-E are isolated, the use of ceftolozane-tazobactam can be considered, after weighing the pros and cons of this approach, to limit exposure to multiple agents and their associated toxicities. However, if this approach is taken, close monitoring of patients for an appropriate clinical response is advised.
Question 2.7: What is the role of non-β-lactam therapy for the treatment of infections caused by Enterobacterales at moderate risk of clinically significant AmpC production due to an inducible ampC gene?
Suggested approach: Nitrofurantoin and TMP-SMX are preferred treatment options for uncomplicated cystitis caused by AmpC-E. Ciprofloxacin, levofloxacin, or an aminoglycoside (as a single dose) are alternative treatment options for AmpC-E uncomplicated cystitis. TMP-SMX, ciprofloxacin, or levofloxacin are preferred treatment options for pyelonephritis or cUTIs caused by AmpC-E.
Aminoglycosides are alternative options for pyelonephritis or cUTI when resistance or toxicities preclude the use of TMP-SMX or fluoroquinolones. For AmpC-E infections outside of the urinary tract, transitioning from cefepime to oral TMP-SMX, ciprofloxacin, or levofloxacin should be considered, if susceptibility is demonstrated.
Rationale
Preferred treatment options for AmpC-E uncomplicated cystitis include nitrofurantoin 19 or TMP- SMX 42,213. Ciprofloxacin, levofloxacin, or a single dose of IV aminoglycosides are alternative are alternative treatment options for AmpC-E uncomplicated cystitis, as described in Question 1.1.
TMP-SMX, ciprofloxacin, or levofloxacin are preferred treatment options for pyelonephritis or cUTIs caused by AmpC-E, as described in Question 1.2. Cefepime is a preferred agent for pyelonephritis or cUTI when resistance or toxicities preclude the use of TMP-SMX or fluoroquinolones. Aminoglycosides are alternative options for the treatment of AmpC-E pyelonephritis or cUTI as discussed in Question 1.2.
The role of TMP-SMX or fluoroquinolones for the treatment of AmpC-E infections outside of the urinary tract has not been formally evaluated in clinical trials. However, neither TMP-SMX nor fluoroquinolones are substrates for AmpC hydrolysis. Transitioning to oral TMP-SMX or fluoroquinolones has been shown to be effective for Enterobacterales bloodstream infections, including those caused by AmpC-E, after appropriate clinical milestones are achieved 73,74. These agents are reasonable treatment options for patients with AmpC-E infections if the conditions described in Question 1.3 are met.
Section 3: Carbapenem-Resistant Enterobacterales
CRE are defined as members of the Enterobacterales order resistant to at least one carbapenem antibiotic (i.e., ertapenem, meropenem, imipenem, doripenem) or producing a carbapenemase enzyme214. Resistance to at least one carbapenem other than imipenem is required for bacteria intrinsically less susceptible to imipenem (e.g., Proteus , Morganella spp., Providencia spp.)214.
CRE comprise a heterogenous group of pathogens encompassing multiple mechanisms of resistance, broadly divided into those that are not carbapenemase-producing and those that are carbapenemase-producing. CRE that are not carbapenemase-producing may be the result of amplification of non-carbapenemase β-lactamase genes (e.g., ESBL genes) with concurrent outer membrane porin disruption215. Carbapenemase-producing isolates account for 35%-83% of CRE cases in the United States, with higher percentages observed when restricting the definition of CRE to require resistance to meropenem or imipenem216-218.
The most common carbapenemases in the United States are K. pneumoniae carbapenemases (KPCs), which are not limited to K. pneumoniae isolates. Other carbapenemases include New Delhi metallo-β-lactamases (NDMs), Verona integron-encoded metallo-β-lactamases (VIMs), imipenem- hydrolyzing metallo-β-lactamases (IMPs), and oxacillinases (e.g., OXA-48-like)218-220. NDM, VIM, and IMP carbapenemases are collectively referred to as metallo-β-lactamases (MBLs)221.
The CDC characterized over 42,000 CRE isolates collected between 2017-2019 and found that approximately 35% of CRE clinical or surveillance isolates in the United States carry one of the main five carbapenemase genes216. Of these carbapenemase-producing isolates, the specific prevalence by carbapenemase gene family was as follows: blaKPC (86%), blaNDM (9%), blaVIM (<1%), blaIMP (1%), or blaOXA- 48-like (4%)216. A more recent cohort of 261 consecutive clinical CRE isolates (defined as resistance to meropenem or imipenem) from 2019-2021 from across the United States found that 83% of isolates were carbapenemase producing (blaKPC [80%], blaNDM [15%], blaIMP [5%], blaOXA-48-like [7%]); between 2019 to 2021 the percentages of blaKPC decreased from 74% to 57%, whereas the percentages of isolates with MBL genes (e.g., blaNDM, blaVIM, blaIMP) increased from 4% to 20% and those with blaOXA-48-like increased from 1% to 8%218.
Knowledge of the carbapenemase produced when CRE is identified in clinical isolates is important in guiding treatment decisions as specific newer β-lactam antibiotics have activity against specific carbapenemases. Phenotypic tests such as the modified carbapenem inactivation method differentiate carbapenemase and non-carbapenemase-producing CRE but generally do not provide information on the specific carbapenemase present16,222. This information is increasingly important given the evolving epidemiology of carbapenemases. Clinical microbiology laboratories are strongly encouraged to implement either nucleic acid or antigen testing to identify the presence of the specific carbapenemases produced by clinical CRE isolates. Treatment suggestions for CRE infections listed below assume that in vitro activity of preferred and alternative antibiotics has been demonstrated.
Question 3.1: What are preferred antibiotics for the treatment of uncomplicated cystitis caused by CRE?
Suggested approach: Nitrofurantoin, TMP-SMX, ciprofloxacin, or levofloxacin are preferred treatment options for uncomplicated cystitis caused by CRE, although the likelihood of susceptibility to any of these agents is low. An aminoglycoside (as a single dose), oral fosfomycin (for E. coli only), colistin, ceftazidime-avibactam, meropenem-vaborbactam, imipenem-cilastatin-relebactam, or cefiderocol, are alternative treatment options for uncomplicated cystitis caused by CRE.
Rationale
Clinical trial data evaluating the efficacy of most preferred agents for uncomplicated CRE cystitis are not available. However, as nitrofurantoin, TMP-SMX, ciprofloxacin, or levofloxacin all achieve high concentrations in urine, they are expected to be effective for uncomplicated CRE cystitis, if the isolate is susceptible5,19,20,22,23.
A single dose of an aminoglycoside is an alternative option for uncomplicated CRE cystitis, for reasons described in Question 1.1. In general, higher percentages of CRE clinical isolates are susceptible to plazomicin compared to other aminoglycosides46. Oral fosfomycin is an alternative treatment option exclusively for uncomplicated cystitis caused by E. coli, including if carbapenem resistant, as discussed in Question 1.119.
Colistin (the active form of the commercially available parenteral inactive prodrug colistimethate sodium) is an alternative agent for uncomplicated CRE cystitis. Colistin converts to its active form in the urinary tract223. Clinicians should remain cognizant of the associated risk of nephrotoxicity. Polymyxin B should not be used as treatment for uncomplicated CRE cystitis, due to its predominantly nonrenal clearance and lower rates of success when compared to aminoglycosides224,225. Ceftazidime-avibactam, meropenem-vaborbactam, imipenem-cilastatin-relebactam, and cefiderocol are alternative options for uncomplicated CRE cystitis131,226-230.
Question 3.2: What are preferred antibiotics for the treatment of pyelonephritis or cUTI caused by CRE?
Suggested approach: TMP-SMX, ciprofloxacin, or levofloxacin are preferred treatment options for pyelonephritis or cUTI caused by CRE, if susceptibility is demonstrated. Ceftazidime-avibactam, meropenem-vaborbactam, imipenem-cilastatin-relebactam, and cefiderocol are also preferred treatment options for pyelonephritis or cUTIs. Aminoglycosides are alternative options for the treatment of pyelonephritis or cUTI caused by CRE.
Rationale
Although the minority of CRE are expected to retain susceptibility to TMP-SMX, ciprofloxacin, or levofloxacin, they are all preferred agents to treat CRE pyelonephritis or cUTI if susceptibility is demonstrated40-42. Ceftazidime-avibactam, meropenem-vaborbactam, imipenem-cilastatin-relebactam, and cefiderocol are preferred treatment options for pyelonephritis and cUTIs caused by CRE based on clinical trials showing non-inferiority of these agents to common comparator agents for UTIs131,226-230. Isolates included in these trials were overwhelmingly carbapenem susceptible.
Aminoglycosides are suggested as alternative agents for the treatment of CRE pyelonephritis or cUTI45,47,48,231, as described in Question 1.2.
Fosfomycin is not suggested for the treatment of pyelonephritis or cUTI given its limited renal parenchymal concentrations. More data are needed to evaluate the role of oral fosfomycin for patients with pyelonephritis or cUTI, particularly when administered as a multidose regimen and after several days of preferred therapy as further described in Question 1.2. Of note, in both clinical trials described in Question 1.2, no patients had CRE infections33,51.
Question 3.3: What are the preferred antibiotics for the treatment for infections caused by CRE outside of the urinary tract that are not carbapenemase producing?
Suggested approach: For infections caused by Enterobacterales isolates that are NOT carbapenemase producing that exhibit susceptibility to meropenem and imipenem (i.e., MICs ≤1 µg/mL), but are not susceptible to ertapenem (i.e., MICs ≥1 µg/mL), the use of extended-infusion meropenem (or imipenem- cilastatin) is suggested. For infections caused by Enterobacterales isolates that are NOT carbapenemase producing and that do not exhibit susceptibility to any carbapenem, ceftazidime-avibactam, meropenem-vaborbactam, and imipenem-cilastatin-relebactam are preferred treatment options.
Rationale
For infections caused by Enterobacterales isolates that are not carbapenemase producing that exhibit susceptibility to meropenem and imipenem (i.e., MICs ≤1 µg/mL), but are not susceptible to ertapenem (i.e., MICs ≥1 µg/mL), extended-infusion meropenem (or imipenem-cilastatin) are suggested (Table 1). An evaluation of CRE isolates submitted to the CDC indicated that less than 3% of the 1,249 isolates resistant to ertapenem but susceptible to meropenem and imipenem contained a carbapenemase gene232. Standard-infusion meropenem or imipenem-cilastatin may be reasonable for uncomplicated cystitis (Table 1).
For isolates that are not carbapenemase producing that are susceptible to meropenem but not susceptible to imipenem (and vice versa), in the absence of data to inform the optimal treatment approach, the panel suggests basing the treatment decision on the severity of illness of the patient and site of infection. For example, in this scenario, meropenem may be a reasonable treatment for a UTI but not for a complex intra-abdominal infection. The panel suggests against the use of meropenem- vaborbactam or imipenem-cilastatin-relebactam to treat ertapenem-resistant, meropenem-susceptible and imipenem-susceptible infections since these agents are unlikely to offer any substantial benefit beyond that of extended-infusion meropenem or imipenem-cilastatin alone.
It was previously considered standard practice to administer extended-infusion meropenem in combination with a second agent, frequently polymyxins or aminoglycosides, for the treatment of infections caused by CRE isolates with meropenem MICs as high as 8-16 µg/mL 233. PK/PD data suggested that extended-infusion meropenem may lead to sufficient drug concentrations for the treatment of infections caused by organisms with carbapenem MICs in this range234-236. However, subsequent observational and trial data indicate increased mortality and excess nephrotoxicity associated with polymyxin or aminoglycoside-based regimens relative to newer β-lactam-β-lactamase inhibitor agents for the treatment of CRE infections 237-251. Therefore, the panel advises against the use of extended-infusion carbapenems with or without the addition of a second agent for the treatment of CRE infections when susceptibility to meropenem or imipenem has not been demonstrated252,253. It is plausible that the addition of vaborbactam or relebactam may decrease MICs of meropenem or imipenem even in isolates without a carbapenemase because of other β-lactamases (e.g., ESBLs) that may be overproduced253.
Tigecycline or eravacycline are alternative options for the treatment of CRE infections not involving the bloodstream or urinary tract (Question 3.8). Their activity is independent of the presence or type of carbapenemase.
Question 3.4: What are the preferred antibiotics for the treatment of infections outside of the urinary tract caused by CRE if KPC production is present?
Suggested approach: Meropenem-vaborbactam, ceftazidime-avibactam, and imipenem-cilastatin- relebactam are preferred treatment options for KPC-producing Enterobacterales infections. Cefiderocol is an alternative option.
Rationale
Preferred agents for KPC-producing infections include meropenem-vaborbactam, ceftazidime- avibactam, or imipenem-cilastatin-relebactam. All three agents appear to have greater than 95% activity against KPC-producing Enterobacterales in the United States254. Although all three are preferred agents for the treatment of KPC-producing infections, the panel slightly favors meropenem-vaborbactam, followed by ceftazidime-avibactam, and then imipenem-cilastatin-relebactam, based on available data regarding clinical outcomes (i.e., fewest clinical data available for imipenem-cilastatin-relebactam) and the likelihood of emergence of resistance (i.e., highest likelihood of emergence of resistance for ceftazidime-avibactam)255. These agents are associated with improved clinical outcomes and reduced toxicity compared to regimens previously used to treat KPC-producing infections, which were often polymyxin or aminoglycoside-based237-246,249-251,256. Clinical trials comparing these agents to each other (i.e., meropenem-vaborbactam vs ceftazidime-avibactam) for the treatment of KPC-producing infections are not available.
An observational study compared the clinical outcomes of patients who received either meropenem-vaborbactam or ceftazidime-avibactam for at least 72 hours for the treatment of CRE infections257. Carbapenemase status was largely not reported. Clinical cure and 30-day mortality between the 26 patients who received meropenem-vaborbactam and 105 patients who received ceftazidime-avibactam were 85% and 61% (limited to patients with isolates exhibiting susceptibility to the agent administered) and 12% and 19%, respectively. Although these differences were not statistically significant, they numerically favor meropenem-vaborbactam. Of patients who experienced recurrent CRE infections, 0% (0 of 3) of patients receiving meropenem-vaborbactam and 20% (3 of 15) of patients receiving ceftazidime-avibactam had subsequent CRE isolates resistant to initial therapy. This study had a number of important limitations: likely selection bias due to its observational nature, relatively small numbers of patients, heterogenous sites of CRE infection, more than half of patients had polymicrobial infections, and more than half of patients received additional antibiotic therapy. These limitations notwithstanding, this study suggests that both meropenem-vaborbactam and ceftazidime- avibactam are reasonable treatment options for KPC-producing infections, although the emergence of resistance may be more common with ceftazidime-avibactam (Question 3.7). At least two groups that have published their clinical experiences with the use of ceftazidime-avibactam and meropenem- vaborbactam for CRE infections, where KPCs were the predominant carbapenemase, and similarly found that patients who received meropenem-vaborbactam had a slightly higher likelihood of clinical cure and survival and a lower risk of emergence of resistance than patients treated with ceftazidime-avibactam258-261.
Limited clinical data are available for imipenem-cilastatin-relebactam for the treatment of KPC- producing Enterobacterales. A clinical trial including patients with infections caused by gram-negative organisms not susceptible to imipenem assigned patients to receive either imipenem-cilastatin- relebactam versus imipenem-cilastatin and colistin 240. Of patients with Enterobacterales infections, 40% (2 of 5 patients) and 100% (2 of 2 patients) experienced a favorable clinical response with imipenem- cilastatin-relebactam and imipenem-cilastatin in combination with colistin, respectively 240. It is difficult to draw meaningful conclusions from these data given the small numbers. However, in vitro activity of imipenem-cilastatin-relebactam against KPC-producing Enterobacterales 262-266, clinical experience with imipenem-cilastatin, and the stability of relebactam as a β-lactamase inhibitor 267 suggest imipenem- cilastatin-relebactam is likely to be effective for KPC-producing Enterobacterales if an organism tests susceptible.
Cefiderocol is suggested as an alternative treatment option for CRE infections outside of the urine. Cefiderocol is a synthetic conjugate composed of a cephalosporin moiety and a siderophore, which binds to iron and facilitates bacterial cell entry using active iron transporters 268. Once inside the periplasmic space, the cephalosporin moiety dissociates from iron and binds primarily to PBP3 to inhibit bacterial cell wall synthesis 269. Over 95% of KPC-producing Enterobacterales test susceptible to cefiderocol270. Robust comparative effectiveness data specifically evaluating the role of cefiderocol for KPC-producing Enterobacterales infections are not available. Cefiderocol is suggested as an alternative agent for treating KPC-producing pathogens due to limited clinical outcomes data and to reserve it for the treatment of infections caused by MBL-producing Enterobacterales or glucose non-fermenting gram-negative organisms.
Tigecycline or eravacycline are alternative options for the treatment of KPC-producing infections not involving the bloodstream or urinary tract (Question 3.9). Their activity is independent of the presence or type of carbapenemases.
Question 3.5: What are the preferred antibiotics for the treatment of infections outside of the urinary tract caused by CRE if NDM or other MBL production is present?
Suggested approach: Ceftazidime-avibactam in combination with aztreonam, or cefiderocol as monotherapy, are preferred treatment options for NDM and other MBL-producing Enterobacterales infections.
Rationale
There is no United States Food and Drug Administration (FDA)-approved beta-lactam/beta- lactamase inhibitor with activity against MBL-producing Enterobacterales, although several promising compounds are in late phases of development or have completed clinical trials. Preferred antibiotic options for NDM-producing Enterobacterales (or other MBLs), include ceftazidime-avibactam plus aztreonam, or cefiderocol monotherapy. NDMs hydrolyze penicillins, cephalosporins, and carbapenems, but not aztreonam. Although aztreonam is not hydrolyzed by NDMs, it can be hydrolyzed by other serine β-lactamases that are often co-produced by NDM-producing isolates (e.g., ESBLs, AmpCs, KPCs, or OXA-48-like enzymes). Avibactam generally remains effective at inhibiting the activity of these other β- lactamases. Extrapolating estimates from aztreonam-avibactam, which is not currently clinically available in the United States, the combination of ceftazidime-avibactam and aztreonam is active against approximately 90% of MBL-producing Enterobacterales isolates271-276. The CLSI has endorsed the use of a broth disk elution method to evaluate the susceptibility of MBL-producing Enterobacterales to the combination of ceftazidime-avibactam/aztreonam16,277.
An observational study of 102 adults with bloodstream infections caused by MBL-producing Enterobacterales (82 were NDM-producing) from 2018-2019 compared the outcomes of 52 patients receiving ceftazidime-avibactam in combination with aztreonam versus 50 patients receiving a combination of other agents, primarily polymyxin or tigecycline-based therapy 278. Thirty-day mortality was 19% for the ceftazidime-avibactam/aztreonam group and 44% for the alternate arm, with a significantly lower risk of mortality associated with the former in a propensity score-matched analysis. Another observational study of MBL-producing Enterobacterales infections (328 were NDM-producing, 58% bloodstream) from 2019-2022 included 215 patients receiving ceftazidime-avibactam/aztreonam, 33 patients receiving cefiderocol, and 26 patients receiving colistin-containing regimens279. Unadjusted 30-day mortality was 22%, 33%, and 50% for the ceftazidime-avibactam/aztreonam, cefiderocol, and colistin-containing regimens, respectively279. Ceftazidime-avibactam/aztreonam was associated with reduced 30-day mortality compared to colistin-based regimens.
Strategies for administering the combination of ceftazidime-avibactam and aztreonam are reviewed in Table 1 and Supplemental Material 280-282. Patients should be monitored closely for elevations in liver enzymes, which was observed in approximately 40% of patients in a phase 1 study 283. In rare situations where cefiderocol or combination therapy with ceftazidime-avibactam and aztreonam is not possible (e.g., allergy or intolerance), combination therapy with aztreonam and meropenem- vaborbactam or imipenem-cilastatin-relebactam can be considered, provided OXA-type carbapenemases are not present284,285. Clinical data investigating this approach are limited286.
A second preferred option for the treatment of NDM and other MBL-producing Enterobacterales is cefiderocol. A cohort of 200 North American and European MBL-producing Enterobacterales from 2014-2019 indicated that approximately 92% of isolates were susceptible to cefiderocol287. However, resistance with NDM-producing Enterobacterales has been described with and without prior cefiderocol exposure and thus susceptibility should be confirmed288-291. A clinical trial including patients with MBL-producing Enterobacterales infections identified clinical cure in 80% (8 of 10) and 0% (0 of 4) of patients receiving cefiderocol versus alternate therapy (primarily polymyxin-based therapy), respectively292. Day 28 mortality occurred in 10% (1 of 10) and 75% (3 of 4) of patients, respectively292. Clinical trial data comparing ceftazidime-avibactam/aztreonam versus cefiderocol are not available and both agents are considered preferred treatment options for MBL-producing Enterobacterales infections.
Tigecycline or eravacycline are alternative options for the treatment of NDM-producing infections not involving the bloodstream or urinary tract (Question 3.9). Their activity is independent of the presence or type of carbapenemases.
Question 3.6: What are the preferred antibiotics for the treatment of infections outside of the urinary tract caused by CRE if OXA-48-like production is present?
Suggested approach: Ceftazidime-avibactam is the preferred treatment option for OXA-48-like- producing Enterobacterales infections. Cefiderocol is an alternative treatment option.
Rationale
If OXA-48-like enzymes are produced by an Enterobacterales clinical isolate, ceftazidime- avibactam293 is preferred and cefiderocol is an alternative option294. More than 95% of OXA-48-like- producing Enterobacterales isolates are susceptible to both ceftazidime-avibactam and cefiderocol270,295. Meropenem-vaborbactam and imipenem-cilastatin-relebactam have limited against OXA-48-like producing isolates because vaborbactam and relebactam are unlikely to inhibit OXA-48-like enzymes and are not suggested, even if susceptible in vitro262,296-298.
Clinical trial data comparing ceftazidime-avibactam versus cefiderocol are not available.
Moreover, limited clinical data investigating the clinical outcomes of patients with OXA-48-like infections treated with either agent are available. An observational study including 171 patients with OXA-48-like- producing Enterobacterales infections treated with ceftazidime-avibactam (without a comparator arm) identified 30-day mortality in 22% of patients299. In an observational study of 76 patients with OXA-48-like-producing Enterobacterales bloodstream infections, 12% and 26% of patients died within 30 days amongst the ceftazidime-avibactam and alternative (e.g., polymyxins) arms, respectively293. In a subgroup analysis of 10 patients with OXA-48-positive Enterobacterales who received cefiderocol therapy in two clinical trials, all were alive at day 28 and seven achieved clinical cure294. Although both ceftazidime-avibactam and cefiderocol are expected to be effective against OXA-48-like-producing infections, cefiderocol is suggested as an alternative agent both because of less published clinical data and to reserve it for the treatment of infections caused by MBL-producing Enterobacterales or glucose non-fermenting gram-negative organisms.
Tigecycline or eravacycline are alternative options for the treatment of OXA-48-like-producing infections not involving the bloodstream or urinary tract (Question 3.8). Their activity is independent of the presence or type of carbapenemases.
Question 3.7: What is the likelihood of the emergence of resistance of CRE isolates to the newer β- lactam agents when used to treat CRE infections?
Suggested approach: The emergence of resistance is a concern with all β-lactam agents used to treat CRE infections. Available data suggest the frequency may be highest for ceftazidime-avibactam.
Rationale
As with any β-lactam agent, treatment with a newer β-lactam for CRE infections (i.e., ceftazidime-avibactam, meropenem-vaborbactam, imipenem-cilastatin-relebactam, or cefiderocol) increases the likelihood that subsequent isolates causing infection will no longer be effectively treated with these agents. The most data on the emergence of resistance of CRE to novel agents focuses on KPC-producing isolates. The emergence of resistance to ceftazidime-avibactam most commonly occurs because of mutations in the blaKPC gene translating to amino acid changes in the KPC carbapenemase and increased hydrolysis of ceftazidime. These changes may result in a restoration of susceptibility to carbapenems, but the clinical significance of this finding is unknown300-321. Changes in outer membrane permeability and efflux systems are the primary drivers of the emergence of resistance to meropenem- vaborbactam 260,309,313,322-328 and imipenem-cilastatin-relebactam 329-331. Increases in blaKPC copy numbers have been associated with resistance to all of these agents332-334.
Diverse mechanisms of resistance of Enterobacterales to cefiderocol have been described335,336 including mutations in the TonB-dependent iron transport system288,290,337-340, amino acid changes in AmpC β-lactamases206,207, and increased NDM expression341. Cefiderocol resistance appears notably higher in ceftazidime-avibactam resistant Enterobacterales isolates compared to ceftazidime-avibactam susceptible isolates (83% vs 7%)342. Increasing reports of amino acid insertions in PBP3, the active binding site of cefiderocol and aztreonam, are being described in NDM-producing E. coli isolates316,343-345 leaving no available β-lactam treatment options. Such reports remain rare in the United States272,291,346. Lastly, there has been increasing recognition of cefiderocol resistance in ceftazidime-avibactam resistant KPC producing Enterobacterales and thus susceptibility should always be confirmed(PMID: 34462816) if used in this setting.
Estimates of the emergence of resistance after clinical exposure to ceftazidime-avibactam and meropenem-vaborbactam are approximately 10-20%241,245,261,304 and <5%257,260,347, respectively. The most data are available for ceftazidime-avibactam, in part because it was the first of the newer β-lactam agents active against CRE to receive FDA approval. Limited data exist on the frequency of emergence of resistance of CRE to imipenem-cilastatin-relebactam and cefiderocol.
It is recommended to repeat AST for the newer β-lactams when a patient previously infected with a CRE presents with a sepsis-like picture suggestive of a new or relapsed infection. Furthermore, if a patient was recently treated with ceftazidime-avibactam and presents with a sepsis-like condition, it is suggested to consider a different novel β-lactam agent at least until culture and AST data are available, particularly if AST results from the previous infection indicate that there are other active β-lactam agents. For example, if a patient with a KPC-producing bloodstream infection received a treatment course of ceftazidime-avibactam one month earlier and presents to medical care with symptoms suggestive of infection, consider administering an agent such as meropenem-vaborbactam until organism and AST results are available.
Question 3.8: What is the role of tetracycline derivatives for the treatment of infections caused by CRE?
Suggested approach: Although β-lactam agents remain preferred treatment options for CRE infections, tigecycline and eravacycline are alternative options when β-lactam agents are either not active or unable to be tolerated. Tetracycline derivatives are not suggested for the treatment of CRE urinary tract infections or bloodstream infections.
Rationale
Tetracycline derivatives function independent of the presence or type of carbapenemase. More specifically, both carbapenemase-producing (e.g., KPC, NDM, OXA-48-like carbapenemases) and non- carbapenemase-producing CRE may test susceptible to these agents348-350. The tetracycline-derivative agents achieve rapid tissue distribution following administration, resulting in limited urine and serum concentrations351. Tetracycline derivatives are not suggested for urinary and bloodstream infections, and in at least one observational study have been associated with increased mortality compared to alternative agents for the treatment of CRE bloodstream infections352. Tigecycline or eravacycline can be considered as alternative options for intra-abdominal infections, skin and soft tissue infections, osteomyelitis, and respiratory infections when optimal dosing is used (Table 1). Nausea and emesis are reported in as many as 20-40% of patients receiving tetracycline-derivatives353-355. Of note, CLSI breakpoints are not available for tigecycline or eravacycline against Enterobacterales, but FDA breakpoints are available356 (Table 2). A hollow fiber model357 and clinical outcomes data352 suggest that tigecycline MICs of ≥0.5 for CRE isolates are associated with poor outcomes.
Tigecycline has more published experience available for the treatment of CRE infections compared with eravacycline358-361. A meta-analysis of 15 clinical trials suggested that tigecycline monotherapy is associated with higher mortality than alternative regimens used for the treatment of pneumonia, not exclusively limited to pneumonia caused by the Enterobacterales362. Subsequent investigations have demonstrated that when high-dose tigecycline is prescribed (200 mg IV as a single dose followed 100 mg IV every 12 hours), mortality differences between tigecycline and comparator agents may no longer be evident363-365. Thus, if tigecycline is prescribed for the treatment of CRE infections, the panel recommends that high-dosages be administered 366 (Table 1).
The clinical relevance of differences in MIC distributions between tigecycline and eravacycline described in some studies is unclear because of differences in the PK/PD profile of these agents367-369. Fewer than five patients with CRE infections were included in clinical trials that investigated the efficacy of eravacycline358,370 and post-marketing clinical reports describing its efficacy for the treatment of CRE infections are limited371.
Minimal clinical data are also available investigating the effectiveness of minocycline against CRE infections372,373, but data suggest a lower proportion of CRE isolates are susceptible to minocycline compared to tigecycline or eravacycline350. The panel suggests using minocycline with caution for the treatment of CRE infections. Data evaluating the activity of omadacycline, a tetracycline-derivative with both an IV and oral formulation, against CRE suggest reduced potency relative to other tetracycline derivatives and an unfavorable PK/PD profile (Question 1.3)374-377. Omadacycline is not suggested for the treatment of CRE infections.
Question 3.9: What is the role of polymyxins for the treatment of infections caused by CRE?
Suggested approach: Polymyxin B and colistin are not suggested for the treatment of infections caused by CRE. Colistin is an alternative agent for uncomplicated CRE cystitis.
Rationale
Observational and clinical data indicate increased mortality and excess nephrotoxicity associated with polymyxin-based regimens relative to comparator agents237-245,251. Concerns about the clinical effectiveness of polymyxins, PK/PD data, and accuracy of polymyxin susceptibility testing led the CLSI to eliminate a susceptible category for colistin and polymyxin B378. The panel suggests that these agents be avoided for the treatment of CRE infections, with the exception of colistin as an alternative agent against CRE cystitis. Polymyxin B should not be used as treatment for CRE cystitis, due to its predominantly nonrenal clearance224.
Question 3.10: What is the role of combination antibiotic therapy for the treatment of infections caused by CRE?
Suggested approach: Combination antibiotic therapy (i.e., the use of a β-lactam agent in combination with an aminoglycoside, fluoroquinolone, tetracycline, or polymyxin) is not suggested for the treatment of infections caused by CRE.
Rationale
Although empiric combination antibiotic therapy increases the likelihood that at least one active therapeutic agent for patients at risk for CRE infections is being administered, data do not indicate that continued combination therapy—once the β-lactam agent has demonstrated in vitro activity—offers any additional benefit379. Rather, the continued use of a second agent increases the likelihood of antibiotic-associated adverse events379. Additionally, clinical data indicating that combination therapy prevents the emergence of resistance are lacking.
Randomized trial data are not available comparing the novel β-lactam agents as monotherapy and as a component of combination therapy (e.g., ceftazidime-avibactam versus ceftazidime-avibactam and tobramycin). The limited observational data available have not identified improved outcomes with combination therapy250,258,299,380. An observational study compared the clinical outcomes of 165 patients receiving ceftazidime-avibactam and 412 patients receiving ceftazidime-avibactam plus a second agent for the treatment of KPC-producing infections258. Thirty-day mortality was essentially identical at approximately 25% in both study arms.
Based on available outcomes data, clinical experience, and known toxicities associated with aminoglycosides, fluoroquinolones, tetracyclines, and polymyxins, the panel does not suggest combination therapy for CRE infections when susceptibility to a preferred β-lactam agent has been demonstrated.
Section 4: Pseudomonas aeruginosa with Difficult-to-Treat Resistance
MDR P. aeruginosa is defined as P. aeruginosa not susceptible to at least one antibiotic in at least three antibiotic classes for which P. aeruginosa susceptibility is generally expected: penicillins, cephalosporins, fluoroquinolones, aminoglycosides, and carbapenems381. In 2018, the concept of “difficult-to-treat” resistance was proposed382. In this guidance document, DTR is defined as P. aeruginosa exhibiting non-susceptibility to all of the following: piperacillin-tazobactam, ceftazidime, cefepime, aztreonam, meropenem, imipenem-cilastatin, ciprofloxacin, and levofloxacin.
MDR P. aeruginosa or DTR P. aeruginosa generally evolve as a result of an interplay of multiple resistance mechanisms, including decreased expression of outer membrane porins (e.g., OprD), increased production of or amino acid substitutions within Pseudomonas-derived cephalosporinase (PDC) enzymes (commonly referred to as pseudomonal AmpC enzymes), upregulation of efflux pumps (e.g., MexAB-OprM), mutations in PBP targets, and the presence of expanded-spectrum β-lactamases (e.g., blaOXA-10)383,384. Carbapenemase production is a relatively uncommon cause of carbapenem resistance in P. aeruginosa isolates in the United States385,386, but is identified in significant portions of carbapenem-resistant P. aeruginosa in other regions of the world (e.g., 69% in Latin America, 57% Asia), commonly due to the presence of blaKPC or blaVIM enzymes385,387-392. These estimates suggest the prevalence of carbapenemase-producing P. aeruginosa will increase in the United States in coming years. There are other β-lactamase enzymes (e.g., Guiana extended-spectrum beta-lactamase [GES], Vietnamese extended-spectrum beta-lactamase [VEB], Pseudomonas extended resistance [PER] enzymes, KPCs, and NDMs] rarely identified in P. aeruginosa isolates from patients in the United States that may confer elevated MICs to β-lactam agents, including some newer β-lactam agents 13,385,393.
Given that carbapenemases are uncommon in P. aeruginosa isolates in the United States, carbapenemase testing for DTR P. aeruginosa is not as critical as carbapenemase testing for CRE clinical isolates in United States hospitals. However, the panel encourages all clinical microbiology laboratories to perform AST for MDR and DTR P. aeruginosa isolates against newer β-lactam agents (i.e., ceftolozane- tazobactam, ceftazidime-avibactam, imipenem-cilastatin-relebactam, and cefiderocol). It is important to understand local DTR P. aeruginosa ASTs to guide empiric antibiotic decisions when AST results are pending. Treatment suggestions for DTR P. aeruginosa infections assume in vitro activity of preferred and alternative antibiotics has been demonstrated.
Question 4.1: What are preferred antibiotics for the treatment of infections caused by MDR P. aeruginosa?
Suggested approach: When P. aeruginosa isolates test susceptible to both traditional non-carbapenem β-lactam agents (i.e., piperacillin-tazobactam, ceftazidime, cefepime, aztreonam) and carbapenems, the former are preferred over carbapenem therapy. For infections caused by P. aeruginosa isolates not susceptible to any carbapenem agent but susceptible to traditional β-lactams, the administration of a traditional agent as high-dose extended-infusion therapy is suggested. For critically ill patients or those with poor source control with P. aeruginosa isolates resistant to carbapenems but susceptible to traditional β-lactams, use of newer β-lactam agents to which the organisms to with P. aeruginosa test susceptible (e.g., ceftolozane-tazobactam, ceftazidime-avibactam, imipenem-cilastatin-relebactam) is also a reasonable treatment approach.
Rationale
In general, when a P. aeruginosa isolate tests susceptible to traditional β-lactam agents (i.e., piperacillin-tazobactam, ceftazidime, cefepime, aztreonam), fluoroquinolones (i.e., ciprofloxacin, levofloxacin), or carbapenems, the panel prefers an agent from the former two groups be prescribed over carbapenem therapy in an attempt to preserve the activity of carbapenems for future, increasingly drug-resistant infections.
P. aeruginosa not susceptible to a carbapenem agent (e.g., meropenem or imipenem-cilastatin MICs ≥4 µg/mL) but susceptible to other traditional β-lactam agents constitute approximately 20% to 60% of carbapenem-resistant P. aeruginosa isolates 394-400. This phenotype is generally due to lack of or limited production of OprD, which normally facilitates entry of carbapenem agents through the outer membrane of P. aeruginosa into the periplasmic space, but not the entry of other β-lactam agents396-399. Comparative effectiveness studies to guide treatment decisions for infections caused by P. aeruginosa resistant to carbapenems but susceptible to traditional non-carbapenem β-lactams are not available. If the isolate is susceptible to a traditional non-carbapenem β-lactam (e.g., cefepime), the panel’s preferred approach is to administer the non-carbapenem agent as high-dose extended-infusion therapy (e.g., cefepime 2 g IV every 8 hours, infused over at least 3 hours)401,402 (Table 1).
An alternative approach is to administer a newer β-lactam agent (e.g., ceftolozane-tazobactam, ceftazidime-avibactam, imipenem-cilastatin-relebactam). This approach is considered an alternative option to preserve the effectiveness of newer β-lactams for future, increasingly AMR infections. However, for critically ill patients or those with poor source control, use of newer β-lactams for P. aeruginosa infections resistant to carbapenems but susceptible to non-carbapenem β-lactams is a reasonable consideration. Regardless of the antibiotic agent administered, patients infected with P. aeruginosa should be closely monitored to ensure clinical improvement as P. aeruginosa exhibits an impressive capacity to iteratively express additional resistance mechanisms while exposed to antibiotic therapy. As an example, an analysis of 767 episodes of P. aeruginosa bacteremia identified the emergence of resistance to traditional β-lactam agents within 30 days with the following likelihood: piperacillin-tazobactam (8%), ceftazidime (12%), meropenem (14%), and imipenem (27%)403. Clinicians are advised to request repeat AST of subsequent clinical MDR P. aeruginosa isolates obtained from the same patient to monitor for the development of resistance.
Question 4.2: Are there differences in percent activity against DTR P. aeruginosa across available β- lactam agents?
Suggested approach: Differences in DTR P. aeruginosa isolates susceptibility percentages to newer β- lactams exist, in part due to regional differences in enzymatic mechanisms of resistance.
Rationale
Ceftolozane-tazobactam, ceftazidime-avibactam, imipenem-cilastatin-relebactam, and cefiderocol are β-lactam antibiotics which may be active against DTR P. aeruginosa clinical isolates. Summarizing United States surveillance data, ceftolozane-tazobactam, ceftazidime-avibactam, imipenem-cilastatin-relebactam, and cefiderocol are active against approximately 90%, 85%, 86%, and 99% of carbapenem-non-susceptible P. aeruginosa isolates270,404, respectively; lower percent susceptibilities are exhibited by isolates from persons with cystic fibrosis 405,406. The panel suggests always obtaining AST results for the four newer β-lactam agents for DTR P. aeruginosa infections to guide treatment decisions.
Regional differences in susceptibility estimates across the newer agents exist, often because of varying prevalence of enzymatic-based resistance mechanisms. For example, as neither ceftolozane- tazobactam, ceftazidime-avibactam, nor imipenem-cilastatin-relebactam have activity against MBL- producing P. aeruginosa (e.g., VIM, NDM enzymes), the percent activity of all of these agents will be reduced in settings where these enzymes are produced by P. aeruginosa (e.g., Latin America, Middle East)385. As ceftolozane-tazobactam remains ineffective against KPC-producing P. aeruginosa, its percent activity will be reduced in regions of the world when KPC enzymes are more commonly produced by P. aeruginosa (e.g., Latin America, China). Similarly, while ceftazidime-avibactam generally remains effective against GES-producing P. aeruginosa, imipenem-relebactam is less effective in the setting of GES enzymes; there will likely be higher percent susceptibility to ceftazidime-avibactam compared with ceftolozane-tazobactam and imipenem-cilastatin-relebactam in areas where GES enzymes are being produced (e.g., Spain)391,392,407.
The heavier side chain of ceftolozane compared to ceftazidime confers enhanced steric hindrance to limit PDC-mediated hydrolysis408,409. Additionally, ceftolozane’s bacterial entry is minimally impacted in settings of porin loss, compared to ceftazidime. Ceftolozane does not rely on an inhibitor to restore susceptibility to an otherwise inactive β-lactam agent (i.e., ceftolozane has independent activity against DTR P. aeruginosa and does not need to rely on its β-lactamase inhibitor to maintain this activity). By definition, neither ceftazidime nor imipenem are active against DTR P. aeruginosa. Avibactam and relebactam expand activity of these agents mainly through inhibition of PDCs127.
The panel does not suggest testing meropenem-vaborbactam activity against DTR P. aeruginosa isolates. Vaborbactam only marginally restores meropenem’s activity against DTR P. aeruginosa391. There are no CLSI or FDA breakpoints for meropenem-vaborbactam against P. aeruginosa. Some P. aeruginosa isolates may appear susceptible to meropenem-vaborbactam but not meropenem, if applying the CLSI meropenem-vaborbactam Enterobacterales susceptible breakpoint of ≤4 µg/mL to P. aeruginosa isolates. This is likely an artifact of meropenem-vaborbactam being standardly administered as 2 grams IV every 8 hours, infused over 3 hours. Meropenem breakpoints (i.e., ≤2 µg/mL) are based on a dosage regimen of 1 gram IV administered every 8 hours, as a 30-minute infusion16. If meropenem is infused as 2 grams IV every 8 hours over 3 hours it would likely achieve a similar likelihood of target attainment as meropenem-vaborbactam (i.e., approximately 8 µg/mL)410.
As discussed in Question 3.4, cefiderocol is composed of a cephalosporin moiety and a siderophore, which facilitates bacterial cell entry using active iron transporters268. Combining data from 1,500 carbapenem-non-susceptible P. aeruginosa isolates in surveillance studies, over 97% of isolates exhibited susceptibility to cefiderocol (i.e., MICs ≤4 µg/mL) 129,209,411-416.
Question 4.3: What are preferred antibiotics for the treatment of uncomplicated cystitis caused by DTR P. aeruginosa?
Suggested approach: Ceftolozane-tazobactam, ceftazidime-avibactam, imipenem-cilastatin-relebactam, and cefiderocol are the preferred treatment options for uncomplicated cystitis caused by DTR P. aeruginosa. Tobramycin or amikacin (as a single dose) are alternative treatment options for uncomplicated cystitis caused by DTR P. aeruginosa.
Rationale
Ceftolozane-tazobactam, ceftazidime-avibactam, imipenem-cilastatin-relebactam, and cefiderocol are preferred treatment options for uncomplicated DTR P. aeruginosa cystitis, based on clinical trials showing non-inferiority of these agents to common comparator agents for the treatment of UTIs131,228-230,417. Data are insufficient to favor one of these agents over others for the treatment of uncomplicated cystitis; available trials generally do not include patients infected by pathogens with DTR phenotypes. The suggested approach for the treatment of uncomplicated cystitis caused by DTR P. aeruginosa isolates confirmed to produce MBL enzymes (e.g., blaVIM) is reviewed in Question 4.6.
A single dose of tobramycin or amikacin is an alternative option for uncomplicated cystitis caused by DTR P. aeruginosa. A single IV dose of tobramycin or amikacin are likely effective for uncomplicated cystitis as aminoglycosides are nearly exclusively eliminated by the renal route in their active form, with minimal toxicity, but robust clinical data are lacking28. As of 2023, there are no longer breakpoints for gentamicin for P. aeruginosa16 (Table 2). Tobramycin breakpoints are available for P. aeruginosa, regardless of source (susceptible ≤1 µg/mL); however, amikacin breakpoints against P. aeruginosa are only available for infections originating from urinary sources (susceptible ≤16 µg/mL)16. Plazomicin has neither CLSI nor FDA breakpoints against P. aeruginosa. Surveillance studies indicate that plazomicin is unlikely to provide any incremental benefit against DTR P. aeruginosa if resistance to all other aminoglycosides is demonstrated418.
Colistin, but not polymyxin B, is an alternate consideration for DTR P. aeruginosa cystitis as it converts to its active form in the urinary tract223. Clinicians should remain cognizant of the associated risk of nephrotoxicity. The panel does not suggest the use of oral fosfomycin for DTR P. aeruginosa cystitis as it may be associated with a high likelihood of clinical failure. This is in part due to the presence of the fosA gene, which is found in the genome of almost all P. aeruginosa isolates31.
Question 4.4: What are preferred antibiotics for the treatment of pyelonephritis or cUTI caused by DTR P. aeruginosa?
Suggested approach: Ceftolozane-tazobactam, ceftazidime-avibactam, imipenem-cilastatin-relebactam, and cefiderocol are preferred treatment options for pyelonephritis or cUTI caused by DTR P. aeruginosa. Once-daily tobramycin or amikacin are alternative agents for the treatment of DTR P. aeruginosa pyelonephritis or cUTI.
Rationale
Ceftolozane-tazobactam, ceftazidime-avibactam, imipenem-cilastatin-relebactam, and cefiderocol are preferred treatment options for DTR P. aeruginosa pyelonephritis and cUTI, based on clinical trials showing non-inferiority of these agents to common comparator agents 131,228-230,417. Data are insufficient to favor one of these agents over the others for the treatment of pyelonephritis or cUTI. Available trials generally do not include patients infected by P. aeruginosa with DTR phenotypes. The suggested approach for the treatment of pyelonephritis and cUTI caused by DTR P. aeruginosa isolates confirmed to produce MBL enzymes (e.g., blaVIM) is reviewed in Question 4.6. Once-daily tobramycin or
amikacin are alternative agents for the treatment of DTR P. aeruginosa pyelonephritis or cUTI419, although there is a duration-dependent risk of nephrotoxicity49,50. They may be helpful for completing treatment courses (e.g., transitioning from another agent for terminal doses) given their prolonged duration of activity in the renal cortex and the convenience of once daily dosing47,48 (Table 1, Supplemental Material). Changes in the aminoglycoside breakpoints that were implemented in 2023 are reviewed in Question 4.3.
Question 4.5: What are preferred antibiotics for the treatment of infections outside of the urinary tract caused by DTR P. aeruginosa?
Suggested approach: Ceftolozane-tazobactam, ceftazidime-avibactam, and imipenem-cilastatin- relebactam are preferred options for the treatment of infections outside of the urinary tract caused by DTR P. aeruginosa. Cefiderocol is an alternative treatment option for infections outside of the urinary tract caused by DTR P. aeruginosa.
Rationale
Ceftolozane-tazobactam, ceftazidime-avibactam, and imipenem-cilastatin-relebactam are preferred options for the treatment of DTR P. aeruginosa infections outside of the urinary tract, based on in vitro activity138,139,141,264,266,329,420-458, observational studies459-464, and clinical trial data131,135,145,240,465-469. The vast majority of patients in clinical trials receiving newer β-lactam agents were not infected with DTR aeruginosa. Clinical trials comparing novel agents to each other (e.g., ceftolozane-tazobactam versus ceftazidime-avibactam) are lacking. Rather, available studies focus on comparing newer β-lactam agents to older agents (e.g., ceftolozane-tazobactam versus polymyxins), and generally focus on MDR P. aeruginosa and not DTR P. aeruginosa. The suggested approach for the treatment of infections outside of the urinary tract caused by DTR P. aeruginosa isolates confirmed to produce MBL enzymes (e.g., blaVIM) is reviewed in Question 4.6.
An observational study including 200 patients with MDR P. aeruginosa infections compared the outcomes of patients receiving ceftolozane-tazobactam versus polymyxin- or aminoglycoside-based therapy459. Favorable clinical outcomes were observed in 81% of patients receiving ceftolozane- tazobactam versus 61% of patients receiving polymyxin- or aminoglycoside-based therapy; this difference achieved statistical significance. Pooled data from five clinical trials explored differences in clinical responses for 95 patients with MDR P. aeruginosa infections receiving ceftazidime-avibactam versus carbapenem-based comparators with a favorable clinical response observed in 57% (32 of 56 patients) versus 54% (21 of 39) of patients in the two treatment arms, respectively470. Only 66% of isolates were susceptible to ceftazidime-avibactam making interpretation of the results challenging470. An observational study compared 100 patients receiving ceftolozane-tazobactam and 100 patients receiving ceftazidime-avibactam with MDR P. aeruginosa and mortality was approximately 40% in both groups471. However, this study had several limitations (e.g., AST not available for all included isolates, 40% received combination therapy, 50% polymicrobial infections, <50% bacteremia or pneumonia, suboptimal ceftolozane-tazobactam dosing).
A clinical trial including 24 patients infected with imipenem-non-susceptible P. aeruginosa identified a favorable clinical response in 81% (13 of 16) of patients receiving imipenem-cilastatin- relebactam compared to 63% (5 of 8) receiving imipenem-cilastatin in combination with colistin240. While not achieving statistical significance, potentially due to the small sample size, the numerical differences suggest improved outcomes with use of imipenem-cilastatin-relebactam over colistin-based regimens.
A clinical trial compared the outcomes of patients with infections due to carbapenem-resistant organisms treated with cefiderocol versus alternative therapy, which largely consisted of polymyxin- based therapy230. The trial included 22 unique patients with 29 carbapenem-resistant P. aeruginosa infections230. Mortality at the end of therapy was 18% in both the cefiderocol and alternative therapy arms for patients infected with P. aeruginosa. This trial suggests that cefiderocol performs as well as polymyxin-based regimens, but may not improve outcomes, as has been observed with some of the newer β-lactam-β-lactamase inhibitors240,459. Observational data suggesting cefiderocol may be reasonable for the treatment of DTR P. aeruginosa infections are limited by small sample sizes and lack of non-cefiderocol treatment arms472,473. The panel suggests cefiderocol as an alternative option when inactivity, intolerance, or unavailability preclude the use of the newer β-lactam-β-lactamase inhibitors.
Question 4.6: What are preferred antibiotics for the treatment of DTR P. aeruginosa that produce metallo-β-lactamase enzymes?
Suggested approach: For patients infected with DTR P. aeruginosa isolates that are MBL-producing, the preferred treatment is cefiderocol.
Rationale
P. aeruginosa producing MBLs remain uncommon in the United States385,386. Such isolates are more common in other regions of the world266,385,392,474-476. DTR P. aeruginosa isolates exhibiting resistance to all available β-lactam-β-lactamase inhibitors (i.e., ceftolozane-tazobactam, ceftazidime- avibactam, and imipenem-cilastatin-relebactam) should raise suspicion for possible MBL production. MBL-producing P. aeruginosa isolates generally remain susceptible to cefiderocol270.
Clinical data on the use of cefiderocol as a treatment for MBL-producing P. aeruginosa are limited. Seven patients with MBL-producing P. aeruginosa infections were included in two cefiderocol clinical trials292. Although limited in vitro data477 and isolated case reports478,479 suggest potential clinical success with the combination of ceftazidime-avibactam and aztreonam for MBL-producing P. aeruginosa infections, this combination appears unlikely to present a meaningful incremental benefit over aztreonam alone for MBL-producing P. aeruginosa infections273,387. Although avibactam may help reduce the effectiveness of PDC enzymes, the multiple other non-enzymatic mechanisms generally present in DTR P. aeruginosa are likely to impede aztreonam’s ability to reach its PBP3 target. Extrapolating data from aztreonam-avibactam, it is anticipated that ceftazidime-avibactam and aztreonam have activity against <10% of MBL-producing P. aeruginosa273.
Question 4.7: What is the likelihood of the emergence of resistance of DTR P. aeruginosa isolates to the newer β-lactam agents when used to treat DTR P. aeruginosa infections?
Suggested approach: The emergence of resistance is a concern with all β-lactams used to treat DTR P. aeruginosa infections. Available data suggest the frequency may be the highest for ceftolozane- tazobactam and ceftazidime-avibactam, although fewer data are available investigating this issue for imipenem-cilstatin-relebactam and cefiderocol.
Rationale
As with most antibiotic agents, treatment of DTR P. aeruginosa with any of the newer β-lactam agents (i.e., ceftolozane-tazobactam, ceftazidime-avibactam, imipenem-cilastatin-relebactam, or cefiderocol) increases the likelihood that subsequent infections will no longer be effectively treated with these agents. The emergence of resistance to ceftolozane-tazobactam most commonly occurs because of amino acid substitutions, insertions, or deletions in PDCs409,423,480-491. These alterations occur most commonly in or adjacent to a particular region of the PDC known as the omega loop. Similarly, acquired resistance of P. aeruginosa to ceftazidime-avibactam is most frequently the result of alterations in PDCs480,482,483,485,488,490-493.
Mechanisms contributing to P. aeruginosa resistance to imipenem-cilastatin-relebactam are less clear and are generally presumed to be related to loss of OprD and overexpression of efflux pumps (e.g., MexAB-OprM and/or MexEF-OprN)329,494,495. A number of diverse mechanisms of P. aeruginosa resistance to cefiderocol have been described336,496 including mutations in the TonB-dependent iron transport system337-339,497, amino acid changes in PDCs, as well as modifications in the PBP3 target497-499.
Based on available data thus far, the emergence of resistance of P. aeruginosa to newer β- lactams appears most evident for ceftolozane-tazobactam and ceftazidime-avibactam. This may be at least in part because these agents have been prescribed more frequently in clinical practice than imipenem-cilastatin-relebactam and cefiderocol500. Cross-resistance between ceftolozane-tazobactam and ceftazidime-avibactam is high because of structural similarities. In a cohort of 28 patients with DTR P. aeruginosa infections treated with ceftolozane-tazobactam and who had a subsequent DTR P. aeruginosa isolate after the start of therapy, the subsequent isolate was no longer susceptible to ceftolozane-tazobactam 50% of the time after a median duration of 15 days of therapy491. Over 80% of patients with index isolates susceptible to ceftazidime-avibactam had subsequent isolates exhibiting resistance to ceftazidime-avibactam after ceftolozane-tazobactam exposure, and in the absence of ceftazidime-avibactam exposure. Another cohort study including 14 patients with index and subsequent P. aeruginosa isolates after ceftolozane-tazobactam described treatment-emergence resistance in 79% of paired isolates490. Both of these single-center experiences likely overestimate the likelihood of emergence of resistance to ceftolozane-tazobactam given that patients who did not have recurrent P. aeruginosa infections (hence, not included in the cohort) may have been less likely to develop ceftolozane-tazobactam resistant P. aeruginosa isolates. Nevertheless, estimates of emergence of resistance to ceftolozane-tazobactam and ceftazidime-avibactam remain concerning.
Limited data on the frequency of emergence of resistance to imipenem-cilastatin-relebactam exist. However, one report identified the emergence of non-susceptibility to this agent in 26% (5 of 19) of patients receiving imipenem-cilastatin-relebactam for the treatment of P. aeruginosa infections494. Of note, across two clinical trials, none of the 31 patients with P. aeruginosa infections treated with imipenem-cilastatin-relebactam developed treatment-emergent resistance240,468.
Similarly, estimates of the frequency of the emergence of resistance of P. aeruginosa to cefiderocol are incomplete but in a clinical trial, 6% (1/17) of P. aeruginosa isolates treated with cefiderocol developed resistance to this agent230. Another study indicated that cross-resistance to cefiderocol occurred in three of 14 (21%) isolates that developed treatment-emergent resistance to ceftolozane-tazobactam496.
The panel suggests always repeating antibiotic susceptibility testing for the four newer β- lactams when a patient previously infected with a DTR P. aeruginosa presents with a sepsis-like picture suggestive of a new or relapsed infection. Furthermore, if a patient was recently treated with ceftolozane-tazobactam or ceftazidime-avibactam and presents to medical care with symptoms of recurrent infection, the panel suggests considering use of imipenem-cilastatin-relebactam or cefiderocol, particularly if one of these agents tested susceptible previously, at least until culture and AST data are available.
Question 4.8: What is the role of combination antibiotic therapy for the treatment of infections caused by DTR P. aeruginosa?
Suggested approach: Combination antibiotic therapy is not suggested for infections caused by DTR P. aeruginosa if susceptibility to ceftolozane-tazobactam, ceftazidime-avibactam, imipenem-cilastatin- relebactam, or cefiderocol has been confirmed.
Rationale
Although empiric combination antibiotic therapy (e.g., the addition of tobramycin to a β-lactam agent) to broaden the likelihood of at least one active agent for patients at risk for DTR P. aeruginosa infections is reasonable, data do not indicate that continued combination therapy—once the β-lactam agent has demonstrated in vitro activity—offers any additional benefit over monotherapy with the β-lactam antibiotic379. Rather, the continued use of a second agent increases the likelihood of antibiotic- associated adverse events379. Additionally, clinical data indicating that combination therapy prevents the emergence of resistance are lacking.
Clinical trials comparing survival with ceftolozane-tazobactam, ceftazidime-avibactam, imipenem-cilastatin-relebactam, or cefiderocol as monotherapy and as a component of combination therapy are not available (e.g., ceftazidime-avibactam versus ceftazidime-avibactam and tobramycin). Observational studies have not identified a survival advantage with combination therapy472,501,502. Based on toxicities associated with aminoglycosides and polymyxins and clinical outcomes data not demonstrating a benefit with the use of combination therapy for P. aeruginosa infections379, the panel does not suggest that combination therapy be routinely administered for DTR P. aeruginosa infections when susceptibility to a β-lactam agent has been demonstrated.
If no β-lactam agent demonstrates activity against DTR P. aeruginosa, tobramycin (if susceptibility is demonstrated) can be considered in combination with either ceftolozane-tazobactam, ceftazidime-avibactam, imipenem-cilastatin-relebactam, or cefiderocol, preferentially selecting the β- lactam agent for which the MIC is closest to its susceptibility breakpoint. For example, if ceftolozane- tazobactam and ceftazidime-avibactam MICs against a DTR P. aeruginosa isolate are both >128/4 µg/mL (highly resistant) and the imipenem-cilastatin-relebactam MIC is 4/4 µg/mL (intermediate category), imipenem-cilastatin-relebactam in combination with tobramycin is favored. Data are lacking demonstrating a benefit to this approach and it should be considered as a last resort. This approach is suggested as it may increase the likelihood that at least one active agent is being included in the treatment regimen.
If tobramycin does not test susceptible, polymyxin B can be considered in combination with a newer β-lactam. Polymyxin B is preferred over colistin for non-urinary tract infections because it is not administered as a prodrug and therefore can achieve more reliable plasma concentrations than colistin and it has a potentially reduced risk of nephrotoxicity, although limitations across studies preclude accurate determination of the differential risk of nephrotoxicity503-508.
Question 4.9: What is the role of nebulized antibiotics for the treatment of respiratory infections caused by DTR P. aeruginosa?
Suggested approach: The panel does not suggest the use of nebulized antibiotics for the treatment of respiratory infections caused by DTR P. aeruginosa.
Rationale
There have been conflicting findings for the clinical effectiveness of nebulized antibiotics for the treatment of gram-negative pneumonia in observational studies 509-536. At least three clinical trials investigated the outcomes of patients with gram-negative ventilator-associated pneumonia comparing nebulized antibiotics versus placebo. All three trials allowed for the use of systemic antibiotics. In brief, one trial compared the outcomes of 100 adults with pneumonia (34% caused by P. aeruginosa) treated with nebulized colistin versus placebo 537; a second trial compared the outcomes of 142 adults with pneumonia (22% caused by P. aeruginosa) treated with nebulized amikacin/fosfomycin versus placebo 538; and the third trial compared the outcomes of 508 adults with pneumonia (32% caused by P. aeruginosa) treated with nebulized amikacin versus placebo 539. None of the three clinical trials demonstrated improved clinical outcomes or a survival benefit with nebulized antibiotics compared with placebo for the treatment of ventilator-associated pneumonia, including in a subgroup analyses of patients with drug-resistant pathogens 537-539. A meta-analysis of 13 trials including 1,733 adults with ventilator-associated pneumonia indicated that the addition of nebulized antibiotics was associated with at least partial resolution of clinical symptoms of infection compared to the control group; however, there was significant heterogeneity among the pathogens involved and the definition of clinical response across studies540. No survival benefit, reduction in intensive care unit length of stay, or reduction in ventilator days was observed in patients receiving nebulized antibiotics540.
Reasons for the lack of clinical benefit with nebulized antibiotics in available trials are unclear. In a PK/PD modeling study, aerosolized delivery of the prodrug of colistin to critically ill patients achieved high active drug levels in epithelial lining fluid of the lungs 541. However, it is likely that nebulized antibiotics do not achieve sufficient penetration and/or distribution throughout lung tissue to exert significant bactericidal activity 542, likely due in part to the use of parenteral formulations not specifically designed for inhalation in suboptimal delivery devices such as jet nebulizers 543,544. Professional societies have expressed conflicting views regarding the role of nebulized antibiotics as adjunctive therapy to IV antibiotics 545-547. The panel suggests against the use of nebulized antibiotics as adjunctive therapy for DTR P. aeruginosa pneumonia due to the lack of benefit observed in clinical trials, concerns regarding unequal distribution in infected lungs, and concerns for respiratory complications such as bronchoconstriction with use of aerosolized antibiotics 548.
Section 5: Carbapenem-Resistant Acinetobacter baumannii
Carbapenem-resistant Acinetobacter baumannii (CRAB) infections pose significant challenges in healthcare settings549,550. In this guidance document, for simplicity, we will use the term “CRAB” as we recognize that most clinical microbiology laboratories may not be able to accurately separate carbapenem-resistant A. baumannii from other species within the baumannii and calcoaceticus complex551.
The management of CRAB infections is difficult for several reasons. First, CRAB is most commonly recovered from respiratory specimens or wounds. It is not always clear if a CRAB isolate recovered in a respiratory or wound culture represents a colonizing organism in medically complex patients who are ill due to underlying host factors (e.g., patients requiring mechanical ventilation, patients with extensive burns), or a true pathogen, leading to uncertainty about the need for antibiotic therapy. For the same reason, it is challenging to determine if poor clinical outcomes with CRAB infections are attributable to suboptimal antibiotic therapy or to underlying host factors.
Second, once A. baumannii exhibits carbapenem resistance, it generally has acquired resistance to most other antibiotics expected to be active against wild-type A. baumannii leaving few remaining therapeutic options. The production of OXA carbapenemases (e.g., OXA-23, OXA-24/40) mediates resistance to β-lactams including carbapenems and sulbactam551,552. CRAB isolates often produce additional serine β-lactamases (e.g., Acinetobacter baumannii-derived cephalosporinases [ADCs]), further limiting the utility of common β-lactam agents. Sulbactam resistance is driven primarily by the presence of β-lactamases, but also via mutations targeting PBPs (i.e., PBP1a/1b, and PBP3)553-555. Aminoglycoside modifying enzymes or 16S rRNA methyltransferases generally preclude aminoglycosides as treatment options for CRAB556-558. Mutations in the chromosomally-encoded quinolone resistance determining regions generally mediate resistance to fluoroquinolones557.
Finally, a clear “standard of care” antibiotic regimen for CRAB infections against which to estimate the effectiveness of various treatment regimens does not exist. Robust comparative effectiveness studies between commonly used agents are limited. Data supporting a prioritization of specific agents with CRAB activity or the additive benefit of commonly used combination regimens for CRAB infections remain incomplete. This guidance document focuses on the treatment of moderate- severe CRAB infections.
Question 5.1: What is the general approach for the treatment of infections caused by CRAB?
Suggested approach: The use of an antibiotic regimen which includes a sulbactam-containing agent is suggested for the treatment of CRAB infections. The preferred regimen is sulbactam-durlobactam in combination with a carbapenem (i.e., imipenem-cilastatin or meropenem). An alternative regimen is high-dose ampicillin-sulbactam (total daily dose of 9 grams of the sulbactam component) in combination with at least one other agent (i.e., polymyxin B, minocycline > tigecycline, or cefiderocol), if sulbactam- durlobactam is not available.
Rationale
The general approach for the treatment of CRAB infections is to administer combination therapy with at least two agents for the treatment of CRAB infections, at least until an appropriate clinical response is observed, given the limited data supporting the effectiveness of any single antibiotic agent. It is also generally suggested that at least one agent in the combination is sulbactam-based. The preferred sulbactam-based agent is sulbactam-durlobactam in combination with either imipenem- cilastatin or meropenem (Question 5.2). An alternative approach, when sulbactam-durlobactam is not available, is the administration of high-dose ampicillin-sulbactam (total daily dose of 9 grams of the sulbactam component) as a component of combination therapy (Question 5.3). Sulbactam’s unique activity against A. baumannii isolates has been observed through in vitro studies559-561, animal models562, and clinical outcomes data563-568, as described in Question 5.2 and Question 5.3. When high-dose sulbactam is administered, combination therapy is suggested even though only one of seven clinical trials found improved clinical outcomes with the use of combination antibiotic therapy for CRAB infections563,569-574 (Question 5.4). Notably, the clinical trial that demonstrated a benefit with combination therapy was the only one that included high-dose ampicillin-sulbactam in the combination therapy arm563. Additional agents that can be considered in combination with high-dose ampicillin-sulbactam include polymyxin B (Question 5.5), minocycline (Question 5.6), tigecycline (Question 5.6), or cefiderocol (Question 5.7). Fosfomycin and rifampin are not suggested as components of combination therapy571,573,574 (Question 5.3, Question 5.9).
As two large clinical trials have not demonstrated a benefit with the use of high-dose extended-infusion carbapenem therapy in combination with colistin for the treatment of CRAB infections569,570, meropenem or imipenem-cilastatin are not suggested as routine components of CRAB therapy, with the notable exception of when they are administered in combination with sulbactam-durlobactam (Question 5.8). Nebulized antibiotics are not suggested as adjunctive therapy for CRAB pneumonia, due to the lack of benefit observed in clinical trials537-539, concerns regarding unequal distribution in infected lungs, and the potential for respiratory complications such as bronchoconstriction542-544,548 (Question 5.10).
Question 5.2: What is the role of sulbactam-durlobactam for the treatment of infections caused by CRAB?
Suggested approach: Sulbactam-durlobactam is a preferred agent for the treatment of CRAB infections and is suggested to be administered in combination with imipenem-cilastatin or meropenem.
Rationale
Sulbactam-durlobactam became FDA-approved in May 2023. Durlobactam is a β-lactamase inhibitor with potent inhibition of class A (e.g., TEM-1), class C (e.g., ADC), and class D beta-lactamases (e.g., OXA-24/40, OXA-23). It does not inhibit class B MBLs (e.g., NDM), which are rarely produced by CRAB isolates in the United States, but are increasingly problematic in other regions of the world (e.g., at least 5% of CRAB isolates in Latin America from 2017-2019 contained a blaNDM and contemporary estimates are likely higher)575,576. Durlobactam reduces the likelihood of sulbactam hydrolysis by binding to and inhibiting class A, C, and D β-lactamases so that sulbactam can successfully reach its PBP targets577. Sulbactam-durlobactam is administered as 1 gram of sulbactam and 1 gram of durlobactam (2 grams total) every 6 hours as a 3-hour infusion578 (Table 1). This dosing strategy achieves PK/PD target attainment goals for greater than 90% of A. baumannii isolates with sulbactam-durlobactam MICs of ≤4/4 µg/mL, the FDA and CLSI breakpoint578.
Sulbactam-durlobactam was investigated in a clinical trial of patients with pneumonia or bloodstream infections caused by A. baumannii568. Patients were randomized to sulbactam-durlobactam or colistin; all patients also received imipenem-cilastatin, dosed as 1 gram of imipenem every 6 hours. There were 125 patients with CRAB infections for whom the primary outcome of 28-day mortality was evaluated. Mortality occurred in 19% (12/63) of patients in the sulbactam-durlobactam group and 32% (20/62) in the colistin group, meeting the pre-specified non-inferiority criteria. Secondary outcomes also favored sulbactam-durlobactam, including clinical cure (62% versus 40%), microbiologic response (68% versus 42%) at the test of cure visit, and a lower risk of nephrotoxicity (13% versus 38%). It is important to note that the comparator arm in this trial (i.e., colistin plus imipenem-cilastatin) is not a preferred treatment regimen for CRAB infections.
The additive clinical benefit of imipenem-cilastatin to sulbactam-durlobactam is unclear. Some studies suggest that the combination of sulbactam-durlobactam and imipenem-cilastatin lowers the MIC of sulbactam-durlobactam by 1-2-fold579-581. The potential benefit may be related to the additional PBPs that are targeted with multiple β-lactams (i.e., sulbactam preferentially binds to PBP1 and PBP3 while imipenem preferentially binds to PBP2)580,582. Both sulbactam and imipenem have an increased likelihood of successfully reaching their PBP targets under the protection of durlobactam. Moreover, its plausible that imipenem serves as a substrate for OXA-carbapenemase-mediated hydrolysis, potentially enabling more sulbactam to reach its PBP targets. In a hollow fiber infection model, the addition of a carbapenem to sulbactam-durlobactam led to enhanced reductions in bacterial growth583. Clinical data investigating the benefit of sulbactam-durlobactam for the treatment of CRAB infections in the absence of imipenem-cilastatin are not available. Based on the available in vitro data, it is suggested that imipenem-cilastatin be administered as adjunctive therapy to sulbactam-durlobactam. Meropenem is likely a reasonable substitute for imipenem-cilastatin given their similar PBP targets580,581. For patients requiring prolonged durations of therapy (e.g., CRAB osteomyelitis) it may be reasonable to discontinue carbapenem therapy after clinical improvement has occurred. Our understanding of mechanisms of resistance to sulbactam-durlobactam will evolve as this agent is increasingly used in clinical practice. Available data suggest high-level resistance of sulbactam- durlobactam is generally a result of MBL enzymes or PBP3 mutants553,579,584. In settings of resistance to sulbactam-durlobactam (i.e., MICs ≥16/4 µg/mL), the panel suggests considering optimally-dosed non- sulbactam based combinations (i.e., cefiderocol, minocycline, tigecycline, polymyxin B) as sulbactam- based therapy is unlikely to be of substantial therapeutic value.
Question 5.3: What is the role of ampicillin-sulbactam for the treatment of infections caused by CRAB?
Suggested approach: High-dose ampicillin-sulbactam, as a component of combination therapy, is suggested as an alternate agent for CRAB. This approach is suggested only when the unavailability of sulbactam-durlobactam precludes its use.
Rationale
As described in Question 5.2, sulbactam is a competitive, irreversible β-lactamase inhibitor that, in high doses, saturates PBP1a/1b and PBP3 of A. baumannii isolates553,585. Sulbactam’s unique activity against A. baumannii isolates has been demonstrated through PK/PD studies559-561,586-591, animal models562,592, and clinical outcomes data563-567. The panel suggests high-dose ampicillin-sulbactam (total daily dose of 9 grams of the sulbactam component) as a component of combination therapy for CRAB infections (Table 1).
A review of available PK/PD data indicate that sulbactam total daily dosing of 9 grams is likely to achieve sufficient fT>MIC (regardless of a 40% or 60% fT>MIC threshold) for A. baumannii isolates with sulbactam MICs of up 16-32 µg/mL (i.e., sulbactam-resistant isolates)593. Of note, durlobactam is a potent inhibitor of class A, C, and D enzymes commonly produced by CRAB584,594, enabling lower doses of sulbactam which can then successfully reach its PBP targets under the protection of durlobactam. Ampicillin-sulbactam does not have the added protection of a durlobactam-like beta-lactamase inhibitor. Ampicillin-sulbactam uses a 2:1 formulation; for example, 3 grams of ampicillin-sulbactam is comprised of 2 grams of ampicillin and 1 gram of sulbactam. Ampicillin-sulbactam total daily dosages of 27 grams (equivalent to 9 grams of sulbactam) as extended or continuous infusions are suggested (e.g., 9 grams [3 grams of sulbactam] IV every 8 hours infused over 4 hours)559,560,563,564,587,595.
Fewer than 50% of CRAB isolates test susceptible to ampicillin-sulbactam596,597. Insufficient data exist to determine if standard-dose ampicillin-sulbactam and high-dose ampicillin-sulbactam have equivalent efficacy for CRAB infections caused by isolates susceptible to ampicillin-sulbactam. The panel favors high-dose ampicillin-sulbactam, given its theoretical benefit of saturating sulbactam’s PBP targets, particularly as significant amounts of the agent will likely be hydrolyzed by β-lactamases prior to reaching its PBP targets and because of potential inaccuracies with commonly used approaches for ampicillin-sulbactam AST testing for CRAB (i.e., “susceptible” may not actually be “susceptible” using AST methods other than reference broth microdilution)598,599.
Two meta-analyses have evaluated observational and clinical trial data for various treatment regimens against CRAB infections566,567. A meta-analysis published in 2021 included 18 studies and 1,835 patients and found that ampicillin-sulbactam (total daily dose of at least 6 grams of the sulbactam component) in combination with a second agent was the most effective regimen to reduce mortality in critically ill patients infected with CRAB566. An earlier meta-analysis published in 2017 included 23 observational studies or clinical trials and 2,118 patients with CRAB infections567. This analysis identified sulbactam as having the greatest impact on reducing mortality when evaluating sulbactam-based, polymyxin-based, or tetracycline-based regimens.
At least five clinical trials evaluating mortality in patients with CRAB infections have included sulbactam in one of the treatment arms600. When comparing the mortality in the colistin-based arm versus the sulbactam-based arm in these trials the results were as follows: 42% versus 33%601, 82% versus 42%602, 63% versus 50%563, 38% versus 17%603, 32% versus 19%568. Although differences in mortality reached statistical significance in only one of these trials602, all demonstrate a numerical reduction in mortality in the sulbactam-based arm, suggesting a potential benefit with the inclusion of sulbactam in the treatment regimen. Evaluating the totality of in vitro, animal, and clinical data, the panel considers high-dose ampicillin-sulbactam, in combination with a second agent, as an alternative option for the treatment of CRAB infections, when sulbactam-durlobactam is not available.
Question 5.4: What is the role of combination antibiotic therapy for the treatment of infections caused by CRAB?
Suggested approach: Combination therapy with at least two agents, whenever possible, is suggested for the treatment of CRAB infections, at least until clinical improvement is observed, because of the limited clinical data supporting any single antibiotic agent.
Rationale
Combination therapy is suggested for the treatment of CRAB infections, even if a single agent demonstrates activity. In situations when prolonged durations of therapy may be needed (e.g., osteomyelitis), step-down therapy to a single active agent can be considered. In vitro and animal studies have had conflicting findings but several investigations indicate increased bacterial killing with various combination regimens 560,604-612. There are many observational studies evaluating the role of combination therapy versus monotherapy for the treatment of CRAB infections with differing results 612- 633. The heterogeneity in patient populations, infectious sources, inclusion of colonizing isolates, variation in antibiotics and dosages used, small numbers, and imbalances between treatment arms makes interpretation of a number of these studies challenging.
At least seven trials have investigated the role of combination therapy for CRAB infections and only one of the seven trials indicated a potential benefit with combination therapy559,563,570-574,634. Of note, because of inconsistent and unclear colistin dosing reported in studies, the panel elected not to report colistin dosing used in individual trials. None of the seven trials that included a polymyxin arm investigated the role of polymyxin B, which has a more favorable PK profile than colistin 224. Only one of these trials included sulbactam in a treatment arm. Below is a summary of the seven trials, a number of which are limited by small sample sizes.
A trial including 210 ICU patients with invasive CRAB infections compared the outcomes of patients receiving colistin alone versus colistin in combination with rifampicin (known in the United States as rifampin) and found no difference in 30-day mortality with 43% mortality in both study arms572. A second trial including 43 patients with CRAB pneumonia also compared colistin monotherapy and colistin in combination with rifampin 573. In hospital mortality was 73% in the colistin group and 62% in the colistin-rifampin group, not reaching statistical A third study randomized nine patients with colistin-resistant A. baumannii (carbapenem susceptibility status not described) and found no difference in 30-day mortality between the colistin and colistin plus rifampin arms (20% versus 33%, respectively) 574.
A fourth trial including patients with a variety of CRAB infections randomized 94 patients to receive colistin alone or colistin with fosfomycin 571. Mortality within 28 days was 57% versus 47% in the colistin monotherapy and colistin-fosfomycin arms, respectively. IV fosfomycin is not currently available in the United States, making the results of this trial of limited relevance to this guidance document.
Two large trials evaluated the role of colistin monotherapy versus colistin in combination with meropenem570,635. In the first study, 312 patients with CRAB bacteremia, pneumonia, or urinary tract infections were randomized to colistin alone versus colistin plus meropenem (2 grams IV every 8 hours as a 3-hour infusion)570. No difference in 28-day mortality (46% versus 52%) were observed between the groups 570. The second trial included 329 patients with drug-resistant A. baumannii bloodstream infections or pneumonia randomized to colistin alone compared to colistin in combination with meropenem (1 gram IV every 8 hours as a 30-minute infusion)635. The 28-day mortality was 46% versus 42% in the colistin monotherapy and combination therapy arms, respectively635. For both trials, the addition of meropenem to colistin did not improve survival in patients with severe CRAB infections.
The seventh trial included 39 CRAB pneumonia patients, with clinical isolates demonstrating susceptibility to both colistin and sulbactam563. Patients were randomized to colistin monotherapy versus colistin in combination with high-dose sulbactam (total daily dose of 8 grams of the sulbactam component) 563. Clinical improvement by day five was observed in 16% and 70% of patients in the colistin versus colistin-sulbactam arms, respectively, achieving statistical significance. 28-day mortality occurred in 63% and 50% of patients, respectively. Investigators were unblinded to treatment assignment. Moreover, patients were allowed to transition to other antibiotics after day five, precluding an accurate comparison of clinical failure or mortality between the groups.
Although only one of seven clinical trials demonstrated any statistically significant benefit with combination therapy for CRAB infections, the panel favors the use of combination therapy for CRAB infections for the following reasons: (1) the vast majority of clinical trials included combinations not generally administered in clinical practice (e.g., colistin and rifampin) making the applicability of trial results limited; (2) except for sulbactam-durlobactam, there is a lack of robust clinical data supporting the treatment of CRAB infections with any single agent demonstrating in vitro activity against CRAB; the use of two agents may increase the likelihood that at least one active agent is being administered; and (3) high bacterial burdens are expected with CRAB infections due to almost universal delays in initiating effective therapy as common empiric antibiotic regimens are generally not active against CRAB. When considering the high mortality associated with CRAB infections, the benefit of using two agents may outweigh the risks. Potential options for consideration as components of combination therapy in addition to high-dose ampicillin-sulbactam include: tetracycline derivatives (with the most experience available for minocycline), polymyxin B, or cefiderocol (Questions 5.3 to 5.6).The decision to preferentially select one agent over another should be based on patient and infection specific factors (e.g., polymyxin B may be less appealing for patients with chronic kidney diseases [Question 5.5], minocycline may be less appealing for bloodstream infections [Question 5.6]).
Question 5.5: What is the role of the polymyxins for the treatment of infections caused by CRAB?
Suggested approach: Polymyxin B can be considered in combination with at least one other agent for the treatment of CRAB infections.
Rationale
The polymyxins, including both colistin and polymyxin B, have reliable in vitro activity against CRAB isolates, with most of the published literature focusing on colistin 560,561. The panel preferentially suggests polymyxin B when considering polymyxin-based regimens, based on its more favorable PK profile than colistin 224,546,636. Colistin is favored for CRAB UTIs, as it converts to its active form in the urinary tract. In comparison, there is minimal excretion of polymyxin B in the urine (4%)3. There is no CLSI susceptible category for the polymyxins against A. baumannii; the benefit of polymyxins is likely diminished when polymyxin MICs are >2 µg/mL637. Due to certain chemical properties of the polymyxins (e.g., poor diffusion through agar, adherence to microtiter plates) obtaining accurate polymyxin MICs is challenging638.
The panel advises against polymyxin monotherapy for the following reasons: First, concentrations of polymyxins in serum achieved with conventional dosing strategies are highly variable and may be inadequate for effective bactericidal activity 224. Similarly, the activity of IV polymyxins in pulmonary epithelial lining fluid is suboptimal and generally does not result in adequate bacterial killing in the lungs 639-641. Second, dosages required to treat systemic infections approach the threshold for nephrotoxicity, making the therapeutic window extremely narrow (i.e., ~2 µg/mL may be required to achieve 1-log10 reduction in bacterial growth, but this is also the threshold associated with nephrotoxicity) 642. Finally, in the largest clinical trials (over 300 patients in each trial) evaluating the role of colistin monotherapy, mortality was relatively high at 46% in both trials569,570.
Question 5.6: What is the role of tetracycline derivatives for the treatment of infections caused by CRAB?
Suggested approach: High-dose minocycline or high-dose tigecycline can be considered in combination with at least one other agent for the treatment of CRAB infections. The panel prefers minocycline over tigecycline because of the long-standing clinical experience with this agent and the availability of CLSI breakpoints.
Rationale
Several tetracycline derivatives have in vitro activity against CRAB including minocycline, tigecycline, and eravacycline643,644. The frequency of the emergence of resistance to these agents by CRAB isolates is not well defined but occurs through drug efflux stemming from overexpression of various RND-type transporters645,646. A general concern with tetracycline derivatives is that they achieve rapid tissue distribution following administration, resulting in limited concentrations in the urine and serum39. Tetracycline derivatives are not suggested as monotherapy for bloodstream infections.
There has been considerable clinical experience with the use of minocycline since its introduction in the 1960s647. It is commercially available in both oral and IV formulations. Data from critically ill patients who received a single 200 mg dose of minocycline was used to develop a population PK model; a dose of 200 mg of IV minocycline administered every 12 hours was predicted to result in a suboptimal PK/PD profile for organisms with MICs >1 µg/mL648. This is important to recognize as the CLSI breakpoints for minocycline against A. baumannii is ≤4 µg/mL16. Caution is advised with the use of minocycline for CRAB isolates with MICs of 2-4 µg/mL where susceptibility might be reported but suboptimal antibiotic concentrations may be present at sites of infection. International surveillance data suggest minocycline is active against approximately 60-80% of CRAB isolates, but this is likely an overestimation given a susceptibility breakpoint of ≤4 µg/mL was applied649,650. Minocycline has not been subjected to rigorous trials for the treatment of CRAB infections, although case series describing its use are available373,651-654. Drawing conclusions on the effectiveness of minocycline from these observational reports is challenging as they have important limitations (e.g., small sample sizes, selection bias, inadequate distinctions between colonization and infection, heterogeneous sites of infection). Despite the limitations of available data, the panel considers minocycline a treatment option for CRAB infections (dosed at 200 mg twice daily either IV or orally) when used as a component of a combination regimen (Table 1).
Tigecycline is a tetracycline derivative only available as an IV formulation. Neither CLSI nor FDA breakpoints are available for tigecycline against A. baumannii isolates; minocycline MICs cannot be used to predict tigecycline MICs as differences in the likelihood of susceptibility across the tetracycline derivatives exist655. Several observational studies and a meta-analysis of 15 trials suggest that tigecycline monotherapy is associated with higher mortality than alternative regimens used for the treatment of pneumonia, not exclusively limited to CRAB pneumonia362,619,656,657. Subsequent investigations have suggested that when high-dose tigecycline is prescribed (200 mg IV as a single dose followed 100 mg IV q12h), mortality differences between tigecycline and comparator agents are no longer evident363-365. Similar to minocycline, efficacy of tigecycline may be limited when MICs are >1 µg/mL based on PK data derived from critically ill patients658. If tigecycline is prescribed for the treatment of CRAB infections, the panel suggests that high doses are used (Table 2). As with minocycline, tigecycline is suggested to be prescribed in combination with at least one additional agent for CRAB infections. Both agents are associated with nausea in 20-50% of patients, and this is likely more common with higher dosages353-355.
Although eravacycline MICs are generally 2- to 8-fold lower than tigecycline MICs against CRAB 655,659,660, the clinical relevance of the differences in MIC distributions between these agents is unclear due to differences in the PK profile of tigecycline and eravacycline. As with tigecycline, no CLSI breakpoints exist for eravacycline. Small numbers of patients with CRAB infections were included in clinical trials investigating the efficacy of eravacycline358,370. Limited post-marketing clinical reports describing its efficacy for the treatment of CRAB infections are available661,662. In an observational study of 93 patients with CRAB pneumonia, eravacycline was associated with longer durations of mechanical ventilation (11 vs 7 days) and higher 30-day mortality (33% vs 15%) compared to alternative regimens662. All four patients with CRAB bloodstream infections receiving eravacycline died. In light of the limited clinical data supporting the use of eravacycline, the panel suggests limiting its use to situations when other agents are either not active, unable to be tolerated or unavailable.
Pre-clinical data evaluating the activity of omadacycline, a tetracycline derivative with both an IV and oral formulation, suggest reduced efficacy against CRAB isolates relative to other tetracycline derivatives. A PK/PD profile suggests omadacycline has very limited activity against CRAB isolates374-377. Clinical data are limited to a small, uncontrolled case series663. The panel does not suggest the use of omadacycline to treat CRAB infections.
Question 5.7: What is the role of cefiderocol therapy for the treatment of infections caused by CRAB?
Suggested approach: Cefiderocol should be limited to the treatment of CRAB infections refractory to other antibiotics or in cases where intolerance or resistance to other agents precludes their use. When cefiderocol is used to treat CRAB infections, the panel suggests prescribing it as part of a combination regimen.
Rationale
Cefiderocol is a cephalosporin conjugated to a siderophore with pre-clinical and clinical data investigating its role against CRAB isolates. International surveillance studies indicate that approximately 95% of CRAB isolates are susceptible to cefiderocol using the CLSI breakpoint of ≤4 µg/mL270,664 (Table 2). Determining CRAB susceptibility to cefiderocol, however, is challenging, in part due to variable iron concentrations in media. Moreover, MIC results are not always reproducible across methods, heteroresistance may be observed, and broth microdilution results can be challenging to interpret as trailing endpoints, haziness, or a paradoxical effect may obscure interpretation665-667. Furthermore, preclinical data suggest higher cefiderocol PK/PD targets needed for A. baumannii are higher than for other gram-negative organisms and bactericidal activity of cefiderocol in animal models of A. baumannii infections has been variable668-671.
A clinical trial including 54 patients with CRAB infections identified mortality at the end of the study to be 49% (19 of 39 patients) versus 18% (3 of 17 patients) in the cefiderocol versus alternative therapy arms (largely composed of polymyxin–based regimens), respectively 230. Poor outcomes with cefiderocol were observed in patients with pneumonia and bloodstream infections. A second trial that included a subgroup of 47 patients with CRAB pneumonia identified 14-day mortality in 22% (5 of 23 patients) versus 17% (4 of 24) of patients in the cefiderocol and meropenem arm, respectively – suggesting outcomes were similar between cefiderocol and a relatively inactive agent 672. Because of the heterogeneity of regimens used in the alternative arms in the first trial and the relatively small numbers of patients with CRAB when combining both trials, contextualizing the results is challenging 268.
In an observational study, 30-day mortality was 34% versus 56% for 124 patients with CRAB infections receiving cefiderocol versus colistin-based regimens, respectively 673. Recurrent CRAB infections, however, were more likely in the cefiderocol arm (17% versus 7%). Among the 8 patients in the cefiderocol group who experienced a recurrent CRAB infection, 50% had subsequent isolates exhibiting resistance to cefiderocol. Additional observational data suggest cefiderocol may be reasonable for the treatment of CRAB infections but these studies are generally limited by small sample sizes, lack of a comparator group or very heterogenous comparator groups, and high percentages of concomitant COVID-19 infections674-676.
Combining available data, the panel suggests that if cefiderocol is prescribed for the treatment of CRAB infections, it should be used with caution and as a component of combination therapy, to increase the likelihood that at least one effective agent is included as part of the treatment regimen. The panel also suggests limiting consideration of cefiderocol for CRAB infections after other regimens have been exhausted.
Question 5.8: What is the role of extended-infusion meropenem or imipenem-cilastatin for the treatment of infections caused by CRAB?
Suggested approach: Meropenem or imipenem-cilastatin are not suggested for the treatment of CRAB infections, with the exception of co-administration with sulbactam-durlobactam.
Rationale
In vitro data suggest that triple-combination therapies consisting of (1) meropenem, ampicillin- sulbactam, and minocycline or (2) meropenem, ampicillin-sulbactam, and polymyxin B may lead to microbiological efficacy against CRAB 559-561. Although at least two observational studies suggest favorable outcomes with the inclusion of carbapenems in 3-drug combinations (i.e., ampicillin-sulbactam, carbapenem, colistin), these studies did not compare outcomes of 3-drug combinations versus 2-drug combinations of ampicillin-sulbactam and colistin637,677. As described in Question 5.4, two randomized trials evaluated the role of colistin monotherapy versus colistin plus meropenem and neither trial demonstrated a benefit with the combination of colistin plus meropenem for the treatment of CRAB infections569,570. A secondary analysis of one of the trials found that improved clinical outcomes were not observed with the combination of colistin and meropenem even when in vitro synergy was present678.
Imipenem-cilastatin may retain activity against some meropenem-resistant isolates679-681; however, MICs of both agents against CRAB isolates are almost always significantly higher than 8 µg/mL570,635. With highly elevated MICs, it appears unlikely that either meropenem or imipenem- cilastatin would offer any incremental benefit when used as a component of combination therapy, with the notable exception of sulbactam-durlobactam (Question 5.2).
Question 5.9: What is the role of the rifamycins for the treatment of infections caused by CRAB?
Suggested approach: Rifampin or other rifamycins are not suggested for the treatment of CRAB infections.
Rationale
The rifamycin class of antibiotics includes agents such as rifampin, rifabutin, and rifapentine that inhibit bacterial RNA polymerase682. Data indicate that rifabutin has potent activity against A. baumannii in both in vitro and animal models, which is significantly greater than that exhibited by rifampin683-685.
Synergy between rifabutin and the polymyxins has been proposed due to the latter’s ability to disrupt bacterial membrane permeability, which may facilitate intracellular penetration of rifamycin and subsequent inhibition of bacterial protein synthesis684.
Three clinical trials compared the clinical outcomes of CRAB-infected patients receiving colistin alone versus colistin in combination with rifampin (Question 5.4)572-574. None of these trials demonstrated a survival benefit with the addition of rifampin. Admittedly, there are limitations to all these trials including suboptimal dosing of colistin and small sample sizes. It is unknown if a clinical benefit would have been observed if rifabutin had been used in place of rifampin686. In light of the known toxicities and drug interactions associated with the rifamycins687 and the absence of a benefit observed in clinical trials, the panel does not favor the use of rifamycins as components of CRAB therapy.
Question 5.10: What is the role of nebulized antibiotics for the treatment of respiratory infections caused by CRAB?
Suggested approach: Nebulized antibiotics are not suggested for the treatment of respiratory infections caused by CRAB.
Rationale
There have been conflicting findings regarding the clinical effectiveness of nebulized antibiotics for the treatment of gram-negative pneumonia in observational studies509-536. At least three randomized trials evaluated the outcomes of patients with gram-negative ventilator-associated pneumonia comparing nebulized antibiotics versus placebo. All three trials allowed for the use of systemic antibiotics, at the discretion of the treating clinician. In brief, one trial compared the outcomes of 100 adults with pneumonia (65% caused by A. baumannii) treated with nebulized colistin versus placebo537; a second trial compared the outcomes of 142 adults with pneumonia (20% caused by A. baumannii) treated with nebulized amikacin/fosfomycin versus placebo538; and the third trial compared the outcomes of 508 adults with pneumonia (29% caused by A. baumannii) treated with nebulized amikacin versus placebo539. None of the three clinical trials demonstrated improved clinical outcomes or a survival benefit with the use of nebulized antibiotics compared with placebo for the treatment of ventilator- associated pneumonia, including in subgroup analyses of drug-resistant pathogens537-539.
A meta-analysis of 13 trials including 1,733 adults with ventilator-associated pneumonia indicated that no survival benefit, reduction in intensive care unit lengths of stay, or reduction in ventilator days was observed in patients receiving nebulized antibiotics540.
Reasons for the lack of clinical benefit in these trials are unclear. In a PK/PD modeling study, aerosolized delivery of the prodrug of colistin to critically ill patients achieved high active drug levels in the epithelial lining fluid of the lungs 541. However, it is likely that nebulized antibiotics do not achieve sufficient penetration and/or distribution throughout lung tissue to exert significant bactericidal activity 542, likely due in part to the use of parenteral formulations not specifically designed for inhalation in suboptimal delivery devices such as jet nebulizers 543,544. Professional societies have expressed conflicting views regarding the role of nebulized antibiotics as adjunctive therapy to IV antibiotics 545-547. The panel suggests against the use of nebulized antibiotics as adjunctive therapy for CRAB pneumonia, due to the lack of benefit observed in clinical trials, concerns regarding unequal distribution in infected lungs, and concerns for respiratory complications such as bronchoconstriction patients receiving aerosolized antibiotics 548.
Section 6: Stenotrophomonas maltophilia
Stenotrophomonas maltophilia is an aerobic, glucose non-fermenting, gram-negative bacillus that is ubiquitous in water environments688. The organism has a long history of changing nomenclatures and a complicated phylogeny689-691. Although generally believed to be less pathogenic than many other nosocomial organisms, S. maltophilia produces biofilm and virulence factors that enable colonization or infection in vulnerable hosts, such as those with underlying lung disease, persons who inject drugs, and people with hematological malignancies692.
S. maltophilia infections pose management challenges similar to those of CRAB infections. First, although S. maltophilia has the potential to cause serious disease, it is often unclear if S. maltophilia represents a colonizing organism or a true pathogen, particularly in patients with underlying pulmonary conditions such as cystic fibrosis or ventilator dependency693-697. S. maltophilia is often recovered as a component of a polymicrobial infection – further complicating decisions on the necessity of targeted S. maltophilia therapy689,698. Importantly, S. maltophilia can be a true pathogen that causes considerable morbidity and mortality, particularly in patients with hematologic malignancies where it can cause hemorrhagic pneumonia or bacteremia699-705.
Second, treatment selection is hampered by antimicrobial resistance genes and gene mutations carried by S. maltophilia isolates689,691,706. An L1 MBL and L2 serine β-lactamase render most conventional β-lactams ineffective against S. maltophilia. L1 hydrolyzes penicillins, cephalosporins, and carbapenems, but not aztreonam. L2 hydrolyzes extended-spectrum cephalosporins and aztreonam689.
S. maltophilia exhibits intrinsic resistance to aminoglycosides via chromosomal aminoglycoside acetyl transferase enzymes707. Furthermore, S. maltophilia can accumulate multidrug efflux pumps that reduce the activity of TMP-SMX, tetracyclines, and fluoroquinolones, and chromosomal Smqnr genes that further reduce the effectiveness of fluoroquinolones708-711.
Third, a “standard of care” antibiotic regimen for S. maltophilia infections against which to compare the effectiveness of various treatment regimens is not evident712. Clinical trials comparing the effectiveness of commonly used agents for S. maltophilia are lacking. Data to prioritize among agents with in vitro activity against S. maltophilia and to determine the additive benefit of commonly used combination therapy regimens remain incomplete.
Lastly, S. maltophilia AST determination is problematic. The CLSI has established breakpoints for six agents against S. maltophilia: cefiderocol, chloramphenicol, levofloxacin, minocycline, ticarcillin- clavulanate, and TMP-SMX. As of 2023, CLSI breakpoints are no longer available for ceftazidime and it is no longer considered an effective treatment option for S. maltophilia16. Ticarcillin-clavulanate manufacturing has been discontinued and chloramphenicol is rarely used in the United States due to significant toxicities713, leaving four agents for which interpretable antibiotic MIC data can be provided to clinicians. Confidence in MIC interpretive criteria for several of the remaining agents is challenged by concerns about the reproducibility of MICs using testing methods commonly employed in clinical laboratories714,715, limited PK/PD data used to inform breakpoints for most agents, and insufficient data to identify correlations between MICs and clinical outcomes. This guidance document focuses on the treatment of moderate-severe S. maltophilia infections.
Question 6.1: What is a general approach for the treatment of infections caused by S. maltophilia?
Suggested approach: Any of two approaches are preferred options for the treatment of S. maltophilia infections: (1) the use of two of the following agents: cefiderocol, minocycline, TMP-SMX, or levofloxacin or (2) the combination of ceftazidime-avibactam and aztreonam.
Rationale
Given that the isolation of S. maltophilia in culture often represents colonization and not infection, it is prudent to carefully distinguish colonization with S. maltophilia from infection to avoid unnecessary antibiotic use. In situations of S. maltophilia infection, either of two approaches are suggested. One option is combination therapy with at least two active agents (i.e., cefiderocol, minocycline, TMP-SMX, or levofloxacin) – listed in order of preference - at least until clinical improvement is observed, primarily because of the limited supportive data for any individual agent (Questions 6.2, 6.4 to 6.6). Alternatively, the combination of ceftazidime-avibactam and aztreonam can be administered, with the acknowledgement that limited clinical data are available supporting this combination (Question 6.3).
Several investigations suggest increased killing of S. maltophilia with combinations agents presumed to have in vitro activity against S. maltophilia including cefiderocol, minocycline, TMP-SMX, and fluoroquinolones, compared to monotherapy716-719. Clinical outcomes data comparing monotherapy and combination therapy are conflicting and limited to observational studies plagued with concerns such as selection bias, small sample sizes, and significant heterogeneity in patient, microbial, and treatment characteristics712,720-722. A multicenter, observational study of 307 patients with S. maltophilia pneumonia found that combination therapy (largely TMP/SMX with either moxifloxacin or levofloxacin) was not associated with reduced overall 30-day mortality overall compared to monotherapy (largely TMP/SMX or moxifloxacin or levofloxacin) but was associated with reduced mortality in immunocompromised patients and in severely ill patients723. As described in Question 6.2, 6.4 to 6.6, there are either concerning PK/PD data or limited clinical data with cefiderocol, minocycline, TMP-SMX, and levofloxacin individually. The panel favors combination therapy for the treatment of S. maltophilia infections, at least until clinical improvement has occurred.
Question 6.2: What is the role of cefiderocol for the treatment of infections caused by S. maltophilia?
Suggested approach: Cefiderocol as a component of combination therapy, at least until clinical improvement is observed, is a preferred agent for the treatment of S. maltophilia infections.
Rationale
Surveillance studies indicate susceptibility of S. maltophilia isolates to cefiderocol approaches 100%, even against isolates resistant to other commonly prescribed agents411,413,664,718,724,725. The CLSI has a susceptible only breakpoint for cefiderocol against S. maltophilia, because of a paucity of S. maltophilia isolates that are not susceptible to cefiderocol16. Of note, although the emergence of resistance of S. maltophilia to cefiderocol has not been described in patient isolates, cefiderocol- resistant S. maltophilia mutants have been identified in in vitro models – the clinical significance of which remains unclear726,727.
Neutropenic thigh and lung animal infection models demonstrate potent activity of cefiderocol against S. maltophilia and indicate that in vivo efficacy against S. maltophilia appears to correlate with in vitro efficacy, using simulated human dosing671,728-730. A neutropenic rabbit S. maltophilia pneumonia model using human simulated dosages of cefiderocol demonstrated that cefiderocol was able to eradicate S. maltophilia in lung tissue, in contrast to TMP-SMX where residual bacteria were present730. Moreover, 87% of cefiderocol treated rabbits survived compared to 25% of TMP-SMX treated rabbits. No untreated rabbits survived.
A clinical trial evaluating the role of cefiderocol for carbapenem-resistant infections included five patients with S. maltophilia infections230,731. All five patients were assigned to the cefiderocol arm, precluding comparisons between treatment regimens. Four out of five patients died. If limiting the analysis to the three patients with S. maltophilia infections without A. baumannii co-infection, two of three patients died. Additional clinical data evaluating the role of cefiderocol for the treatment of S. maltophilia infections are limited to case reports but several indicate favorable outcomes after failing traditional regimens732-737. Despite the limited availability of clinical data, PK/PD data738 and animal models671,728-730 are very encouraging for the use of cefiderocol in treating S. maltophilia infections. Data are not available to guide the decision to use cefiderocol as a component of combination therapy or as monotherapy. Given the limited clinical experiences with cefiderocol for the treatment of S. maltophilia infections, the panel suggests cefiderocol be considered as a component of combination therapy at least until clinical improvement is observed.
Question 6.3: What is the role of ceftazidime-avibactam and aztreonam for the treatment of infections caused by S. maltophilia?
Suggested approach: Ceftazidime-avibactam and aztreonam is a preferred treatment combination for S. maltophilia infections.
Rationale
The combination of ceftazidime-avibactam and aztreonam (which mimics aztreonam-avibactam) can be used to overcome the activity of both the L1 and L2 β-lactamases intrinsic to S. maltophilia691,739- 744. The L1 MBL hydrolyzes ceftazidime but not aztreonam. The L2 serine β-lactamase hydrolyzes ceftazidime and aztreonam but is inactivated by avibactam. Therefore, the combination of ceftazidime- avibactam and aztreonam enables aztreonam to bypass inactivation and successfully reach its target PBPs of S. maltophilia. Surveillance data indicate aztreonam-avibactam is active against approximately 92% of S. maltophilia isolates739,740,742,743. Despite limited available clinical data with this regimen for the treatment of S. maltophilia infections741,745-747, the combination of ceftazidime-avibactam and aztreonam280,282 is a preferred treatment option for S. maltophilia infections. Strategies for administering the combination of ceftazidime-avibactam and aztreonam are reviewed in Table 1 and Supplemental Material280-282. Patients should be monitored closely for elevations in liver enzymes, which was observed in approximately 40% of patients in a phase 1 study283. The CLSI has endorsed the use of a broth disk elution method to evaluate the susceptibility of S. maltophilia isolates to the combination of ceftazidime-avibactam and aztreonam16,277.
Question 6.4: What is the role of tetracycline derivatives for the treatment of infections caused by S. maltophilia?
Suggested approach: High-dose minocycline, as a component of combination therapy, is an option for the treatment of S. maltophilia infections.
Rationale
Surveillance studies report that minocycline has activity against approximately 70-90% of S. maltophilia isolates719,748-750. These data were generated using minocycline susceptibility breakpoints of ≤4 µg/mL. In 2023, the CLSI lowered the minocycline breakpoints from ≤4 µg/mL to ≤1 µg/mL for S. maltophilia16 (Table 2), and the proportion of S. maltophilia isolates susceptible to minocycline will be reduced. Among the tetracycline derivatives, CLSI breakpoints are only available for minocycline16. Minocycline dosages of 200 mg IV every 12 hours have a >90% probability of achieving PK/PD targets associated with bacterial stasis in a neutropenic mouse thigh model for organisms with MICs of 1 µg/mL, but only a 50% probability of achieving targets associated with 1-log kill751.
Clinical outcomes data investigating the role of tetracycline derivatives for the treatment of S. maltophilia infections are challenging to interpret. Several observational studies have been conducted but are limited by small sample sizes, use of standard-dose and not high-dose minocycline or tigecycline, lack of a comparator arm, heterogeneity in sites of infection, or use of additional antibiotic agents752-756. Studies that included a comparator arm did not indicate any clear failure signals with tetracycline derivatives compared to TMP-SMX or fluoroquinolones.
Despite limitations with interpreting available clinical data, the panel considers high-dose minocycline as a treatment option for S. maltophilia infections, when administered as a component of combination therapy. Because of the slightly more favorable in vitro data with minocycline, more favorable PK/PD data, oral formulation, and potentially improved tolerability of minocycline relative to tigecycline, the panel favors minocycline.
In vitro and in vivo data on the role of eravacycline against S. maltophilia are scarce.
Omadacycline, a tetracycline derivative with oral and IV formulations, has limited in vitro activity against S. maltophilia relative to other tetracycline derivatives748. The panel does not suggest the use of eravacycline or omadacycline for the treatment of S. maltophilia infections.
A general concern with tetracycline derivatives is that they achieve rapid tissue distribution following administration, resulting in limited concentrations in the urine and serum39. Therefore, they are not suggested for S. maltophilia UTIs and should be used with caution and as a component of combination therapy for the treatment of bloodstream infections. Nausea and emesis are reported in as many as 20-40% of patients receiving minocycline or tigecycline353-355.
Question 6.5: What is the role of trimethoprim-sulfamethoxazole for the treatment of infections caused by S. maltophilia?
Suggested approach: TMP-SMX, as a component of combination therapy, is an option for the treatment of S. maltophilia infections.
Rationale
Surveillance studies have consistently shown that TMP-SMX has more than a 90% likelihood of in vitro activity against S. maltophilia757,758, although there is an increasing recognition of S. maltophilia isolates resistant to TMP-SMX718,757,759,760. Despite the longstanding clinical experience with use of TMP- SMX for S. maltophilia infections, several PK/PD studies have emerged indicating that TMP-SMX is not bactericidal against S. maltophilia, even those with low TMP MICs, regardless of the TMP-SMX dosage716,718,719,761,762. At best, TMP-SMX may have the potential to achieve stasis against S. maltophilia.
This is in contrast to organisms like E. coli where at least a one-log kill can be observed in the presence of TMP-SMX at similar exposures761. Some in vitro studies suggest stasis – and possibly even a 1-log kill- can be more reliably achieved when TMP-SMX is administered as a component of combination therapy716,719.
In a neutropenic rabbit lung S. maltophilia model, 5 mg/kg dose twice daily reduced the burden of S. maltophilia in lung tissue, but did not eradicate the bacteria. In contrast, cefiderocol achieved complete bacterial clearance730. Moreover, only 25% of rabbits receiving TMP-SMX survived, compared to 87% receiving cefiderocol.
Rigorous clinical data investigating the effectiveness of TMP-SMX for S. maltophilia infections are lacking. An observational study of 1,581 patients with S. maltophilia identified in respiratory or blood cultures and treated with TMP-SMX or levofloxacin monotherapy was undertaken using an administrative database763. This work suggested that TMP-SMX therapy may be associated with increased mortality compared to levofloxacin in patients with S. maltophilia recovered from respiratory cultures and that TMP-SMX therapy was associated with prolonged hospitalizations. However, there are significant limitations to this study making its findings challenging to interpret (e.g., wide study interval [2005-2017] during which many changes in clinical practice likely occurred, inability to distinguish colonization and infection, inability to adjust for source control, incomplete AST data, inclusion of polymicrobial infections, residual confounding by indication). Given these limitations, the applicability to guide clinical practice is unclear.
Prior to the publication of this work, the largest study evaluating TMP-SMX treatment was a case series of 91 patients with S. maltophilia bloodstream infections, in whom mortality was 25% within 14 days722. The small number of patients in the study who received an agent other than TMP-SMX precluded a comparative effectiveness evaluation. Several relatively small observational studies comparing TMP-SMX and other agents (namely tetracycline derivatives or fluoroquinolones) have been undertaken and generally demonstrated similar outcomes between treatment agents752,754,764-769.
Given the toxicity of TMP-SMX (e.g., hypersensitivity, hyperkalemia, myelosuppression, nephrotoxicity), no established dose-response relationship761, the absence of clinical evidence supporting any particular dose, and evidence that TMP dosing of >15 mg/kg/day may lead to an increased risk of adverse events without any incremental clinical benefit770, a dose range of 10-15 mg/kg (trimethoprim component) of TMP/SMX is suggested for patients with S. maltophilia infections (Table 1). Doses between 10-15 mg/kg/day should provide bacteriostasis for the majority of susceptible isolates. TMP-SMX is a treatment option for S. maltophilia infections, when used in combination with a second agent.
Question 6.6: What is the role of fluoroquinolones for the treatment of infections caused by S. maltophilia?
Suggested approach: Levofloxacin, as a component of combination therapy, is an option for the treatment of S. maltophilia infections.
Rationale
S. maltophilia isolates frequently harbor Smqnr resistance determinants that interfere with fluoroquinolone binding to gyrase and topoisomerase, leading to increased fluoroquinolone MICs691,708. Fluoroquinolone MICs may increase further as a result of overexpression of multidrug-resistant efflux pumps757,771-773. Baseline susceptibility percentages of S. maltophilia to levofloxacin vary from approximately 30% to 80% in surveillance studies718,719,774,775. Several studies have shown that S. maltophilia isolates that test susceptible to levofloxacin can develop elevated levofloxacin MICs during therapy765,766,768,776. CLSI breakpoints exist for levofloxacin against S. maltophilia, but not for ciprofloxacin or moxifloxacin16. In 2023, the CLSI added a comment next to the levofloxacin breakpoint stating “levofloxacin should not be used alone for antimicrobial therapy” for S. maltophilia infections16.
Time-kill curves evaluating ciprofloxacin, levofloxacin, and moxifloxacin monotherapy generally indicate that these agents are inadequate at sustaining inhibition of S. maltophilia growth749,777-780, but suggest that levofloxacin and moxifloxacin may have sufficient activity as components of combination therapy718,719. PK/PD modeling data suggest that fluoroquinolone monotherapy may be insufficient to achieve appropriate target attainment for S. maltophilia infections, even when administered at high dosages749. Neutropenic mouse models suggest that even 750 mg of IV levofloxacin daily may not reliably achieve PK/PD targets associated with bacterial stasis or 1-log killing against a substantial proportion S. maltophilia isolates that have MIC values within the wild-type distribution781. Levofloxacin and moxifloxacin were both associated with improved survival (50%) compared to placebo (0%) in a mouse model of hemorrhagic S. maltophilia pneumonia782. Taken together, these data suggest that fluoroquinolones may not provide sufficient benefit as monotherapy but may provide some additive value when administered as a component of combination therapy.
Clinical data evaluating fluoroquinolones for the treatment of S. maltophilia clinical infections mostly focus on levofloxacin. A meta-analysis including 663 patients from 14 observational studies compared mortality between fluoroquinolones and TMP-SMX, with approximately 50% of patients receiving fluoroquinolones (including, ciprofloxacin [34%] and levofloxacin [57%]) and 50% receiving TMP-SMX764. When pooling the fluoroquinolones, they appeared to be marginally significant in protecting against mortality compared to TMP-SMX, with mortality reported in 26% versus 33% of patients, respectively. When limiting the analysis to patients with S. maltophilia bloodstream infections, where distinguishing colonization and infection is less problematic, a benefit with fluoroquinolone use was not evident.
As discussed in Question 6.4, an observational study of 1,581 patients with S. maltophilia identified in respiratory or blood cultures and treated with TMP-SMX or levofloxacin was undertaken using an administrative database763. Although this work suggested that levofloxacin may be protective against mortality in patients with S. maltophilia recovered from respiratory cultures and marginally protective against mortality regardless of the culture site, there are limitations to this study making its findings challenging to interpret.
Several observational studies comparing fluoroquinolones to other agents (i.e., TMP-SMX, tigecycline) did not identify increased clinical failure signals in the fluoroquinolone arm749,759,763. There are several limitations to these studies including selection bias, small sample sizes, heterogeneity in host and microbial data, and the use of additional active agents.
Due to suboptimal results with fluoroquinolone monotherapy in in vitro studies, known mechanisms of resistance of S. maltophilia to fluoroquinolones, relatively low probability of achieving systemic exposures that correlate with stasis or 1-log kill in animal models, the emergence of resistance during therapy, and inherent biases in observational data, the panel suggests levofloxacin be used as a component of combination therapy, when prescribed for the treatment of S. maltophilia infections. Because of the absence of breakpoints for ciprofloxacin and moxifloxacin, the panel suggests preferentially administering levofloxacin amongst the fluoroquinolones. Adverse events related to fluoroquinolone use and the potential for the emergence of resistant S. maltophilia isolates during levofloxacin therapy should be considered when prescribing this agent783.
Question 6.7: What is the role of ceftazidime for the treatment of infections caused by S. maltophilia?
Suggested approach: Ceftazidime is not a suggested treatment option for S. maltophilia infections due to the presence of β-lactamase genes intrinsic to S. maltophilia that are expected to render ceftazidime inactive. As of 2024, CLSI breakpoints for S. maltophilia to ceftazidime are no longer available.
Rationale
The panel does not suggest prescribing ceftazidime for the treatment of S. maltophilia infections, as intrinsic L1 and L2 β-lactamases are expected to render it ineffective. In vitro models suggest ceftazidime is unable to substantially prevent S. maltophilia growth719. Comparative effectiveness studies evaluating the role of ceftazidime against S. maltophilia infections are virtually non-existent784. As of 2024, the CLSI no longer has susceptibility breakpoints for ceftazidime against S. maltophilia.
Conclusions
The field of AMR is dynamic and rapidly evolving, and the treatment of AMR infections will continue to challenge clinicians. As newer antibiotics against resistant pathogens are incorporated into clinical practice, we are learning more about their effectiveness and propensity to resistance. This treatment guidance will be updated approximately annually and is available at: https://www.idsociety.org/practice-guideline/amr-guidance/.
Notes
Acknowledgement
The expert panel sincerely thank Cornelius J. Clancy, Samuel L. Aitken, and David van Duin for all their contributions to earlier versions of this guidance document. This guidance would have been impossible without their help.
Conflict of Interest Summary
The following list includes what has been reported to IDSA. To provide thorough transparency, IDSA requires full disclosure of all relationships, regardless of relevancy to the guidance topic. Evaluation of such relationships as potential conflicts of interest is determined by a review process which includes assessment by the Board of Directors liaison to the Standards and Practice Guidelines Committee and, if necessary, the Conflicts of Interest and Ethics Committee. The assessment of disclosed relationships for possible conflicts of interests is based on the relative weight of the financial relationship (i.e., monetary amount) and the relevance of the relationship (i.e., the degree to which an association might reasonably be interpreted by an independent observer as related to the topic or recommendation of consideration). IDSA requests panel members to disclose activities and financial relationships/investments related to consultant/advisory roles, promotional speakers bureau, stocks/bonds, honoraria, expert testimony, ownership interest, research grants, organizational benefits, intellectual property, other numeration, activities with other organizations, and relevant financial interest of family members. Readers of this guidance should be mindful of this when the list of disclosures is reviewed.
P.D.T. reports no disclosures. E.L.H. serves as a scientific consultant for Lexi-Comp; serves as an associate editor for the Clinical Infectious Diseases journal; receives research funding from the Maryland Department of Health. J.J. serves as scientific advisor for Shionogi; receives research funding from the Society of Infectious Diseases Pharmacists; served as scientific advisor for Gilead Sciences; received honoraria from Clinical Care Options; owned stock in Vaxart. A.J.M. serves as a scientific advisor for Cepheid, DayZero Diagnostics, and OpGen; receives research funding from the Centers for Disease Control and Prevention, Food and Drug Administration, and National Institute of Health; provided expert testimony sponsored by BioMerieux Inc for the French Embassy. M.J. S. receives research funding from Merck, bioMéreiux, SNIPRBiome, Selux Diagnostics; receives renumeration from AbbVie; serves on a Data and Safety Monitoring Board for AbbVie; received consulting fees from Shionogi and has served on a Data and Safety Monitoring Board for Spero Therapeutics. R.A.B. receives research funding from National Institute of Allergy and Infectious Diseases, Centers for Disease Control and Prevention, Veterans Affairs, and Veterans Health Administration Merit Award; serves as a clinical scientist investigator for Veterans Health Administration Merit Award; receives research funding from Venatorx Pharmaceuticals; received research funding from Shionogi, Merck, Entasis Therapeutics, Wockhardt, Allecra Therapeutics, AstraZeneca, Harrington Family Foundation, Tetraphase Pharmaceuticals, Steris, and Melinta Therapeutics; received an honorarium from Unilab.
All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Conflicts that the editors consider relevant to the content of the manuscript have been disclosed.
References
For a complete list of references, please click here.
Supplemental Material
Publication Disclaimer
© IDSA <2024>. All rights reserved. No part of this publication may be reproduced, stored in a retrieval system, transmitted, used for text and data mining, or used for training artificial intelligence, in any form or by any means, without the prior permission in writing, or as expressly permitted by law, by license or under terms agreed with the appropriate reprographics rights organization. For commercial re-use, please contact reprints@oup.com for reprints and translation rights for reprints. All other permissions can be obtained through the Oxford University Press RightsLink service via the Permissions link for this paper in Clinical Infectious Diseases. For further information please contact journals.permissions@oup.com.